

Evidence of a causal effect of genetic tendency to gain muscle mass on uterine leiomyomata
- PMID: 36726022
- PMCID: PMC9892568
- DOI: 10.1038/s41467-023-35974-7
Evidence of a causal effect of genetic tendency to gain muscle mass on uterine leiomyomata
Abstract
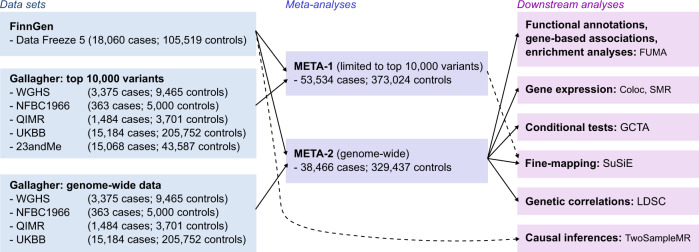
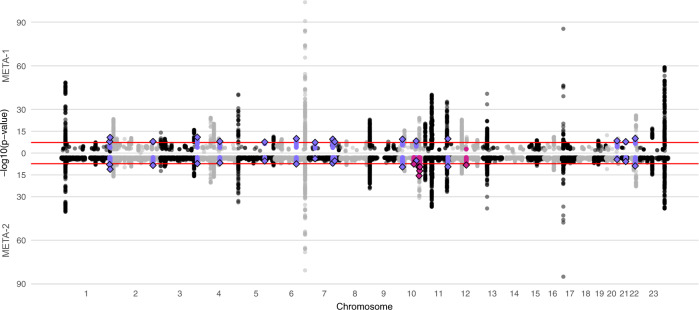
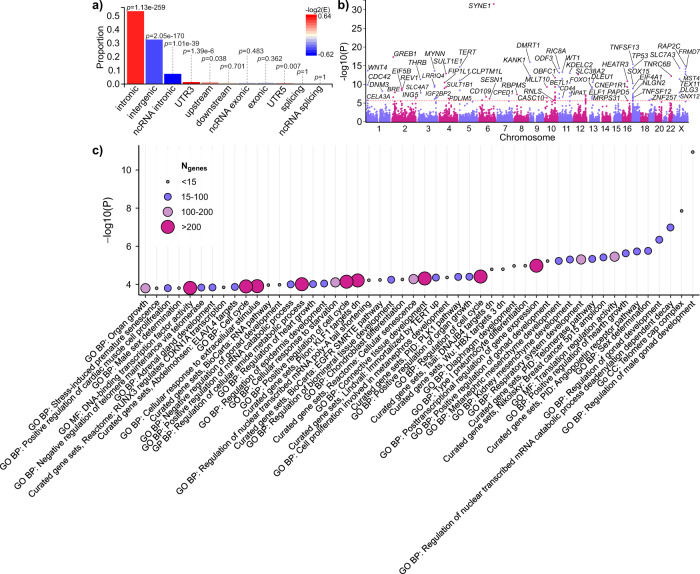
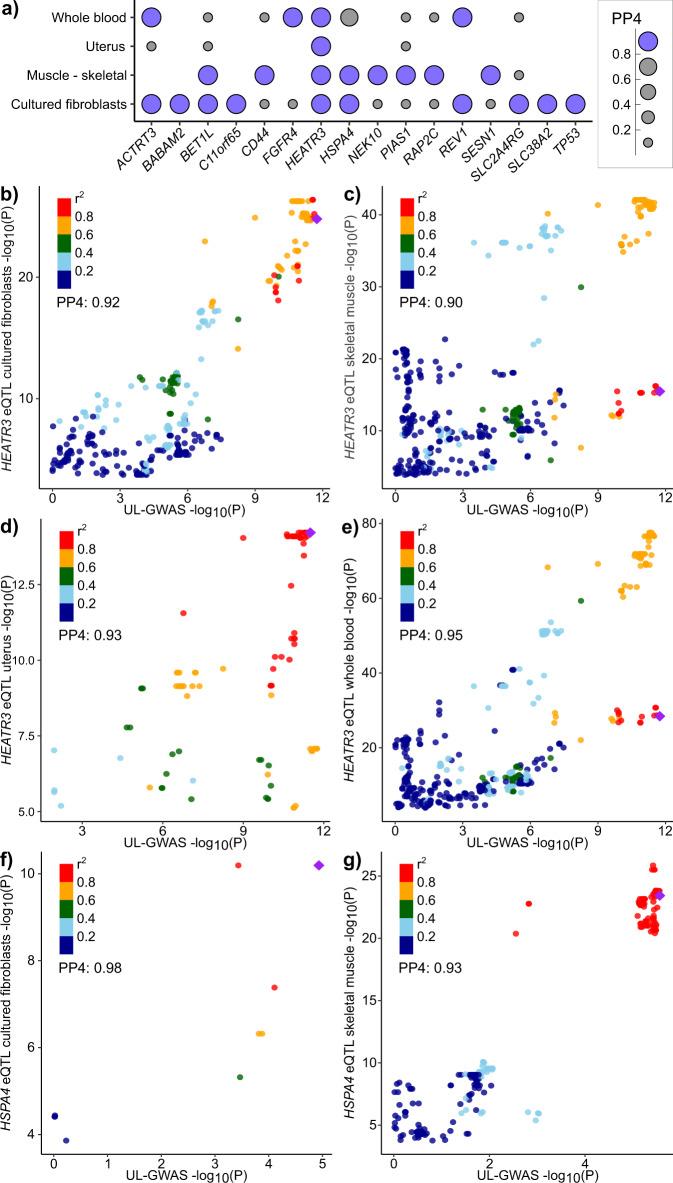
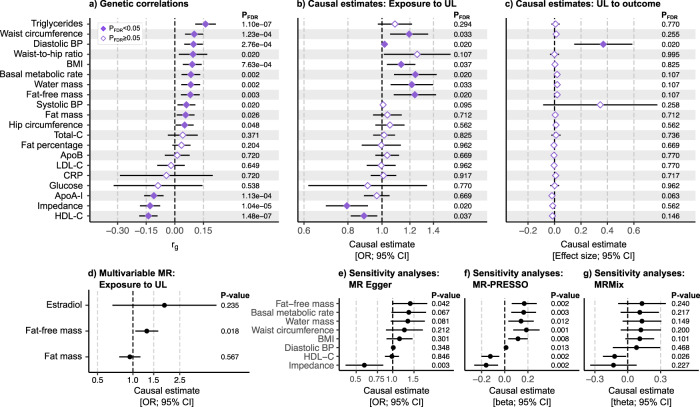
Uterine leiomyomata (UL) are the most common tumours of the female genital tract and the primary cause of surgical removal of the uterus. Genetic factors contribute to UL susceptibility. To add understanding to the heritable genetic risk factors, we conduct a genome-wide association study (GWAS) of UL in up to 426,558 European women from FinnGen and a previous UL meta-GWAS. In addition to the 50 known UL loci, we identify 22 loci that have not been associated with UL in prior studies. UL-associated loci harbour genes enriched for development, growth, and cellular senescence. Of particular interest are the smooth muscle cell differentiation and proliferation-regulating genes functioning on the myocardin-cyclin dependent kinase inhibitor 1 A pathway. Our results further suggest that genetic predisposition to increased fat-free mass may be causally related to higher UL risk, underscoring the involvement of altered muscle tissue biology in UL pathophysiology. Overall, our findings add to the understanding of the genetic pathways underlying UL, which may aid in developing novel therapeutics.
© 2023. The Author(s).
Conflict of interest statement
K.T.Z.: Competing financial interests: Scientific collaborations (grant funding) with Bayer AG, Roche Diagnostics Inc, MDNA Life Sciences, and Evotec. Competing non-financial interests: Board memberships of the World Endometriosis Society, World Endometriosis Research Foundation, and research advisory committee member of Wellbeing of Women UK. CMB: Competing financial interests: Scientific collaborations (grant funding) with Bayer AG, Roche Diagnostics Inc, MDNA Life Sciences, and Evotec. Scientific board Myovant; IDDM Member ObsEva. Competing non-financial interests: Chair ESHRE Endometriosis Guideline Development Group. The remaining authors declare no competing interests.
Figures
References
-
- Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: Ultrasound evidence. Am. J. Obstet. Gynecol. 2003;188:100–107. - PubMed
-
- Walker CL, Stewart EA. Uterine fibroids: the elephant in the room. Science. 2005;308:1589–1592. - PubMed
-
- Stewart, E. A. et al. Uterine fibroids. Nat. Rev. Dis. Prim. 2, 16043 (2016). - PubMed
-
- Mäkinen N, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011;334:252–255. - PubMed
-
- Luoto R, et al. Heritability and risk factors of uterine fibroids - The Finnish Twin Cohort Study. Maturitas. 2000;37:15–26. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
