

High-depth sequencing characterization of viral dynamics across tissues in fatal COVID-19 reveals compartmentalized infection
- PMID: 36732505
- PMCID: PMC9894515
- DOI: 10.1038/s41467-022-34256-y
High-depth sequencing characterization of viral dynamics across tissues in fatal COVID-19 reveals compartmentalized infection
Abstract
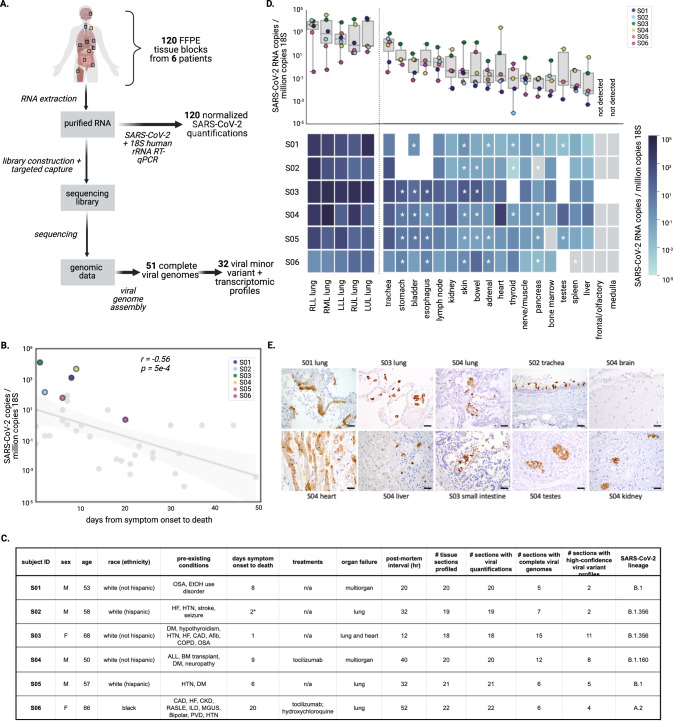
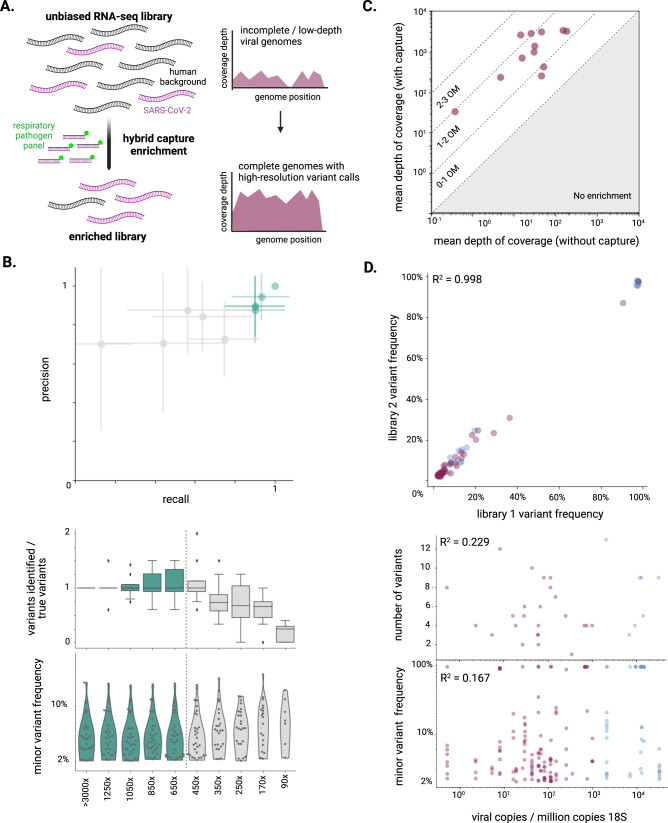
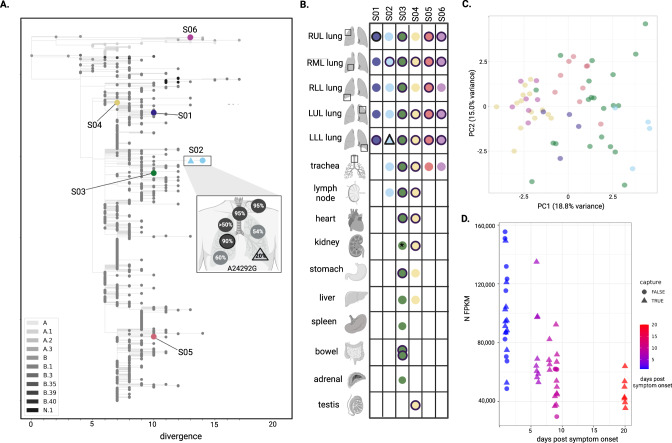
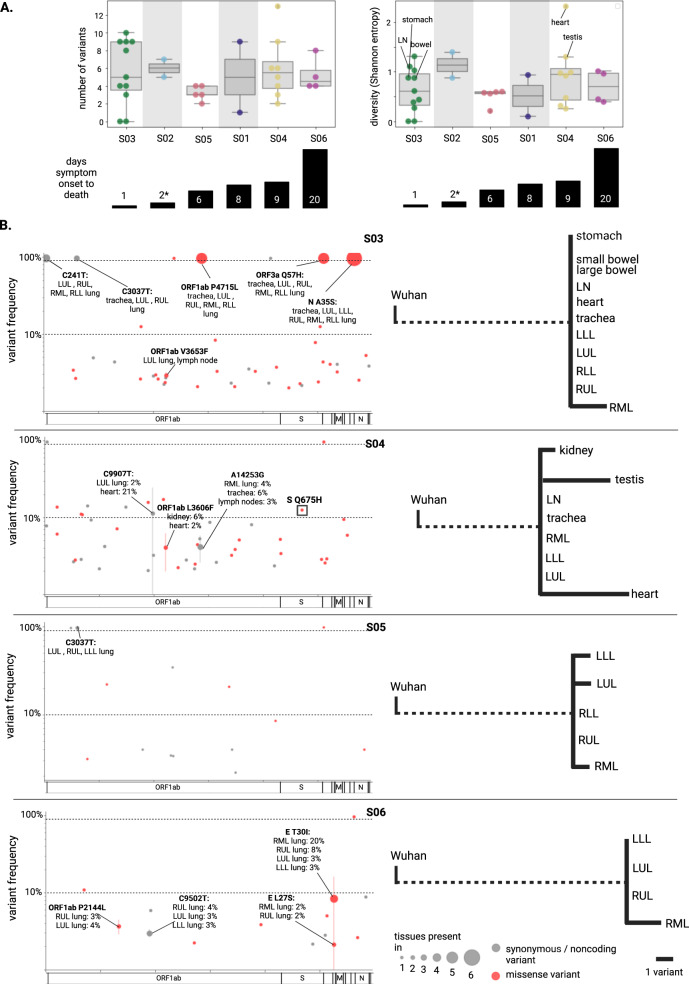
SARS-CoV-2 distribution and circulation dynamics are not well understood due to challenges in assessing genomic data from tissue samples. We develop experimental and computational workflows for high-depth viral sequencing and high-resolution genomic analyses from formalin-fixed, paraffin-embedded tissues and apply them to 120 specimens from six subjects with fatal COVID-19. To varying degrees, viral RNA is present in extrapulmonary tissues from all subjects. The majority of the 180 viral variants identified within subjects are unique to individual tissue samples. We find more high-frequency (>10%) minor variants in subjects with a longer disease course, with one subject harboring ten such variants, exclusively in extrapulmonary tissues. One tissue-specific high-frequency variant was a nonsynonymous mutation in the furin-cleavage site of the spike protein. Our findings suggest adaptation and/or compartmentalized infection, illuminating the basis of extrapulmonary COVID-19 symptoms and potential for viral reservoirs, and have broad utility for investigating human pathogens.
© 2023. The Author(s).
Conflict of interest statement
P.C.S. is a co-founder and consultant at Sherlock Biosciences Inc. and Delve Bio, and is a Board Member of Danaher Corporation; she holds equity in all three companies. She has several patents related to diagnostics, genome sequencing, and informatics, including patents licensed to Sherlock Biosciences. All other authors declare no competing interests.
Figures
References
-
- Topol EJ. COVID-19 can affect the heart. Science. 2020;370:408–409. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
