

Landscape of pathogenic mutations in premature ovarian insufficiency
- PMID: 36732629
- PMCID: PMC9941050
- DOI: 10.1038/s41591-022-02194-3
Landscape of pathogenic mutations in premature ovarian insufficiency
Abstract
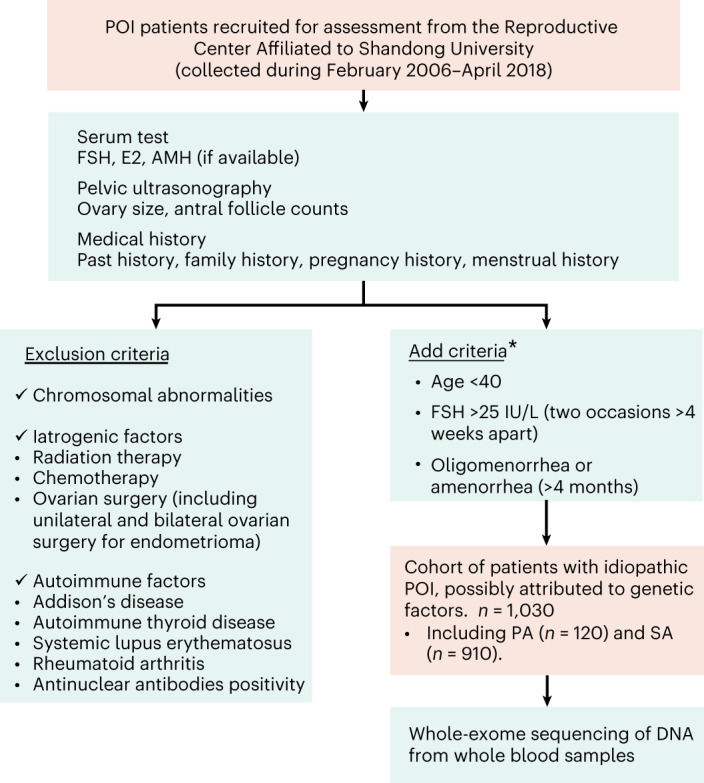
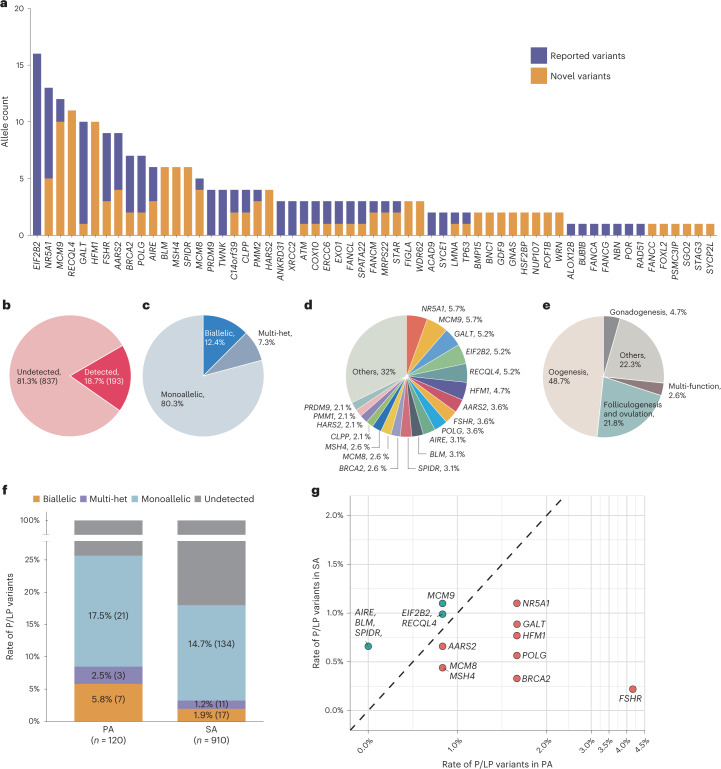
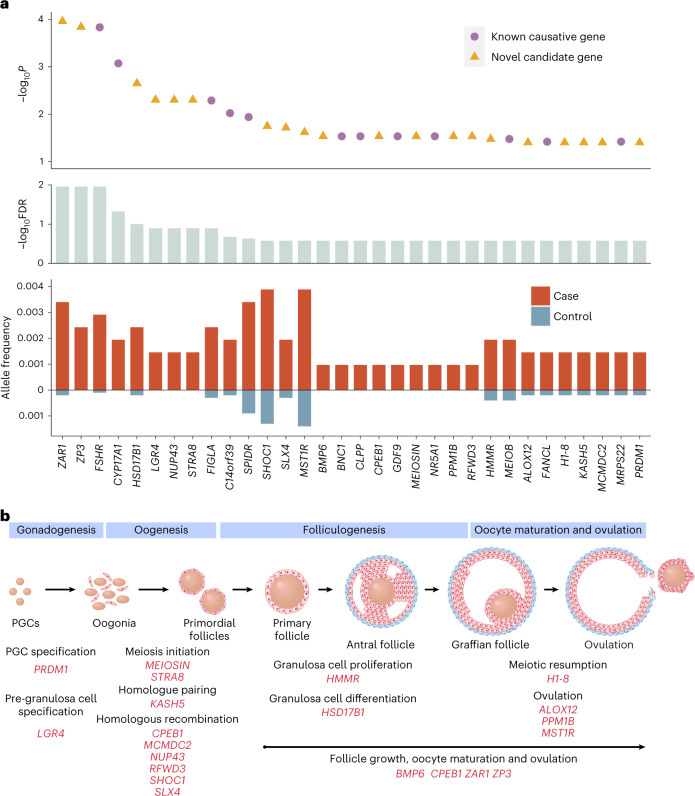
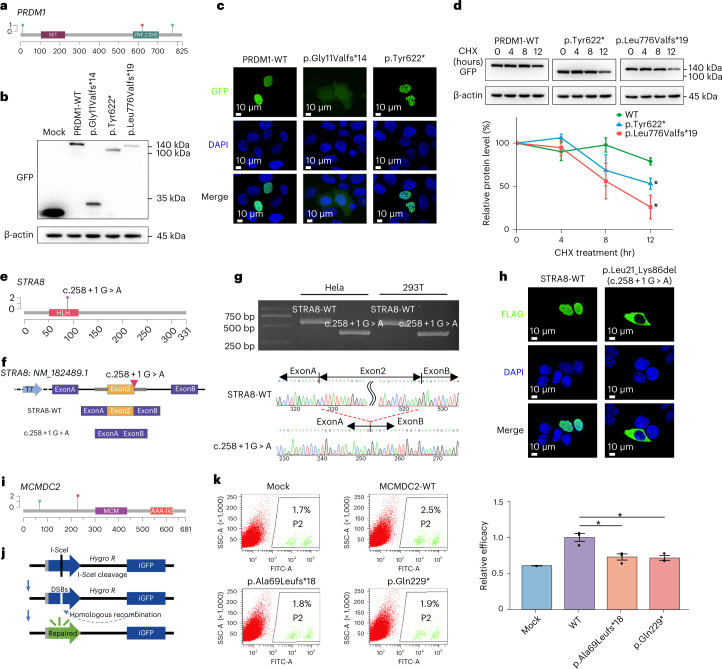
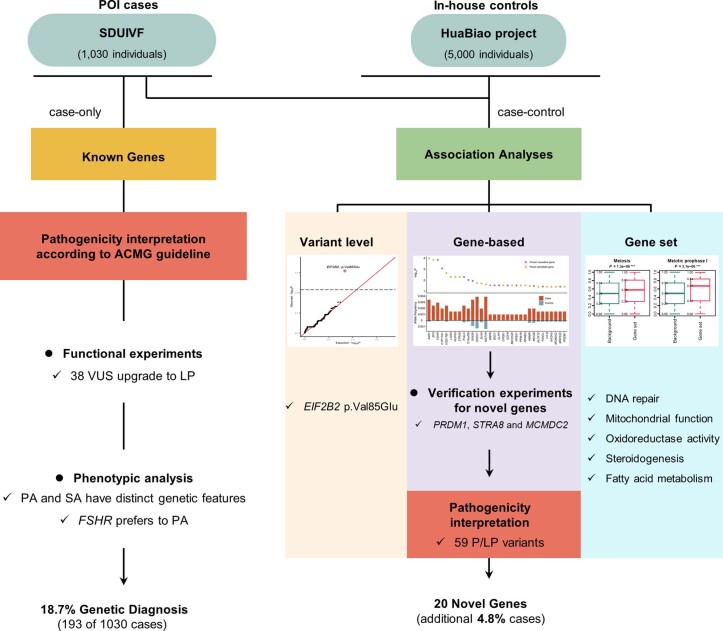
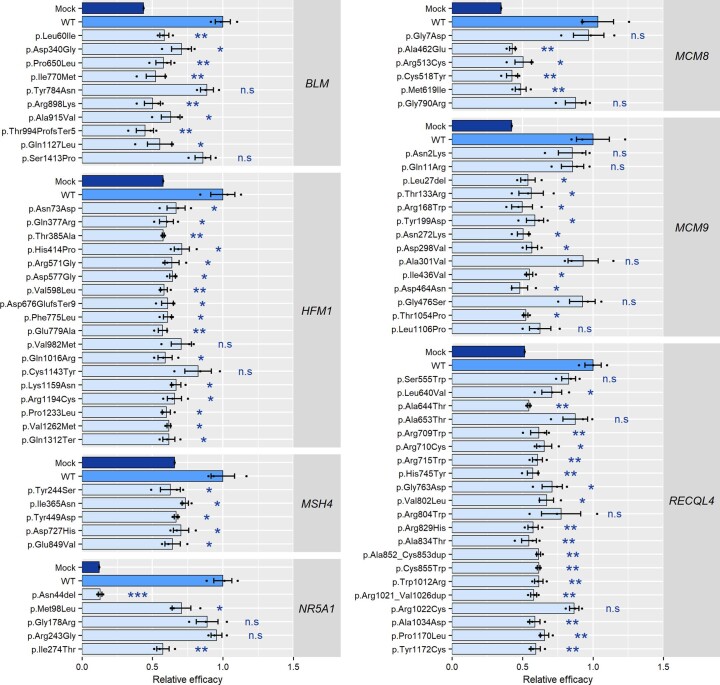
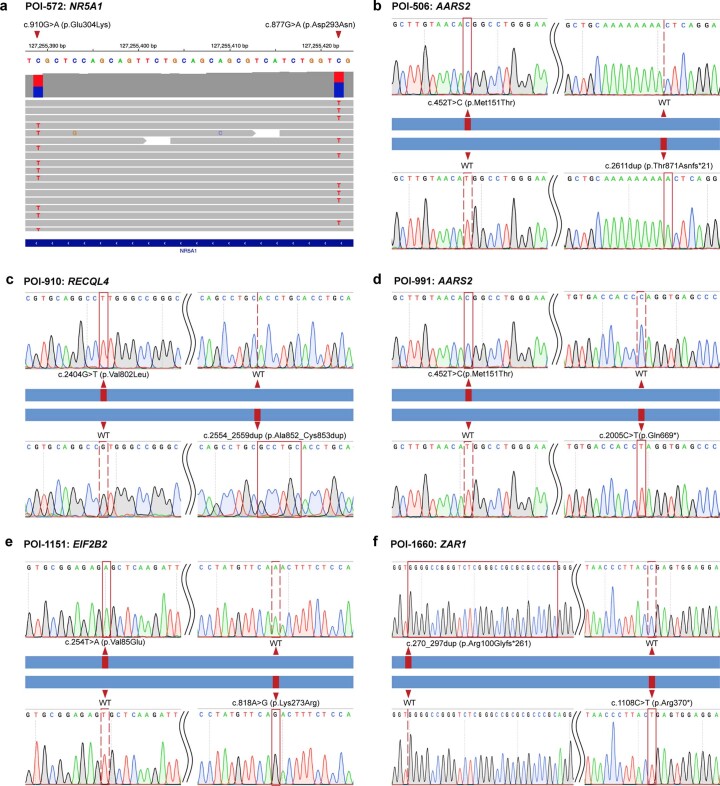
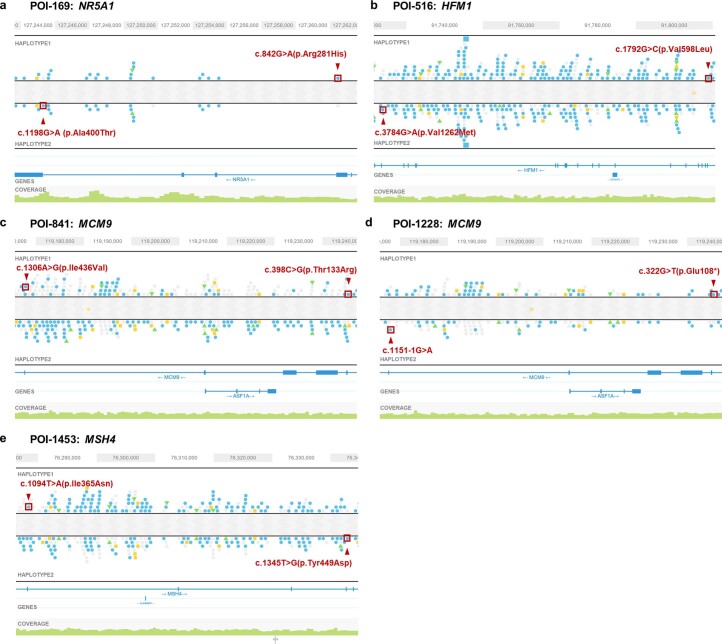
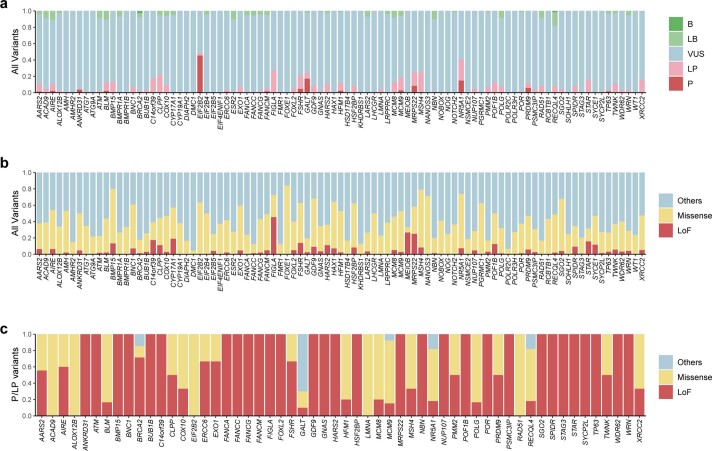
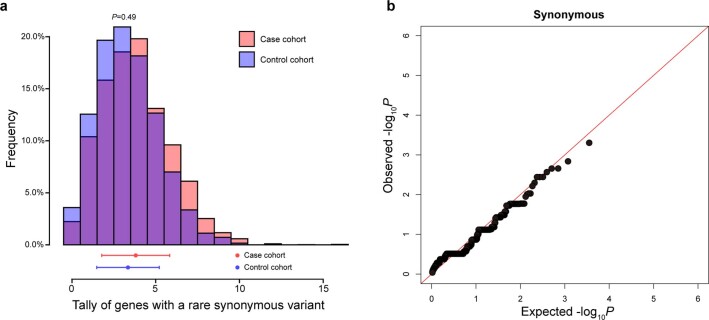
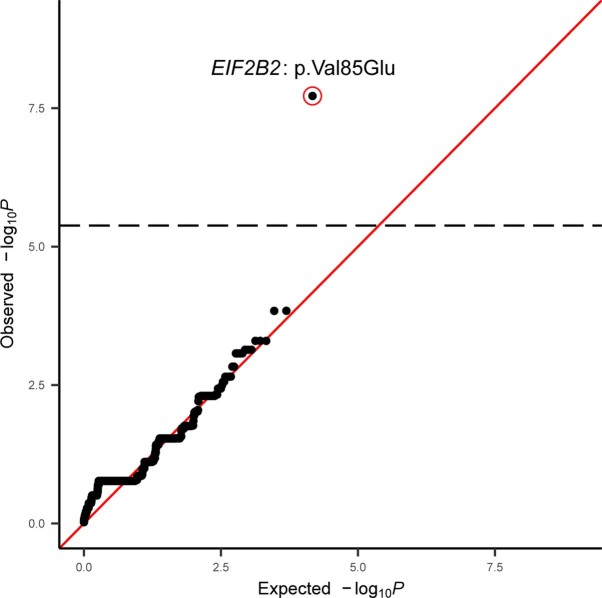
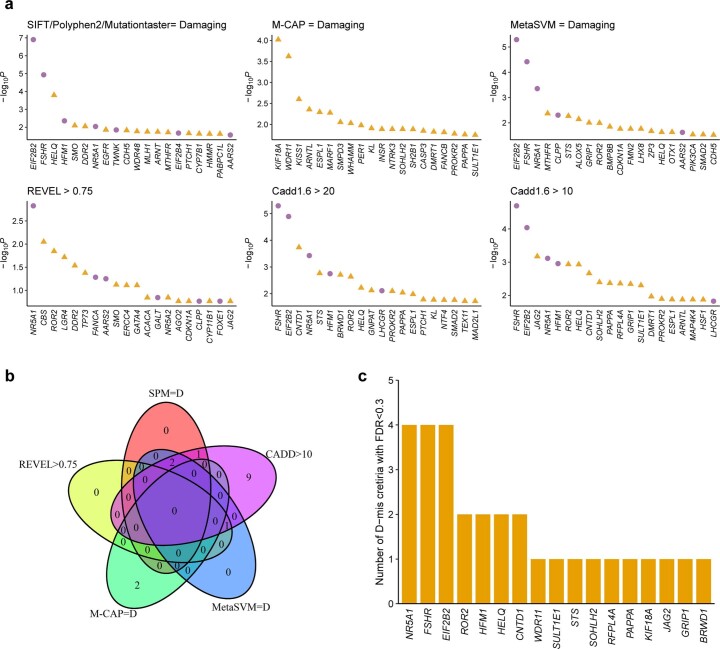
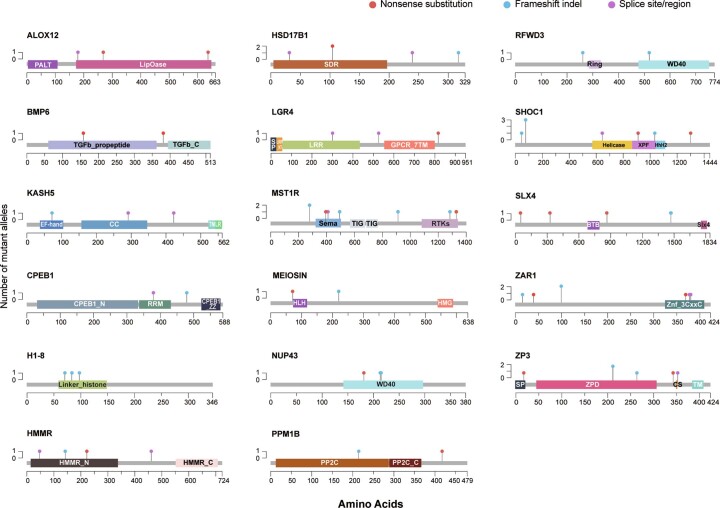
Premature ovarian insufficiency (POI) is a major cause of female infertility due to early loss of ovarian function. POI is a heterogeneous condition, and its molecular etiology is unclear. To identify genetic variants associated with POI, here we performed whole-exome sequencing in a cohort of 1,030 patients with POI. We detected 195 pathogenic/likely pathogenic variants in 59 known POI-causative genes, accounting for 193 (18.7%) cases. Association analyses comparing the POI cohort with a control cohort of 5,000 individuals without POI identified 20 further POI-associated genes with a significantly higher burden of loss-of-function variants. Functional annotations of these novel 20 genes indicated their involvement in ovarian development and function, including gonadogenesis (LGR4 and PRDM1), meiosis (CPEB1, KASH5, MCMDC2, MEIOSIN, NUP43, RFWD3, SHOC1, SLX4 and STRA8) and folliculogenesis and ovulation (ALOX12, BMP6, H1-8, HMMR, HSD17B1, MST1R, PPM1B, ZAR1 and ZP3). Cumulatively, pathogenic and likely pathogenic variants in known POI-causative and novel POI-associated genes contributed to 242 (23.5%) cases. Further genotype-phenotype correlation analyses indicated that genetic contribution was higher in cases with primary amenorrhea compared to that in cases with secondary amenorrhea. This study expands understanding of the genetic landscape underlying POI and presents insights that have the potential to improve the utility of diagnostic genetic screenings.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


Comment in
-
Identification of pathogenic DNA variants in premature ovarian insufficiency.Nat Med. 2023 Feb;29(2):315-316. doi: 10.1038/s41591-022-02196-1. Nat Med. 2023. PMID: 36792852 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
