

Biologically informed deep learning to query gene programs in single-cell atlases
- PMID: 36732632
- PMCID: PMC9928587
- DOI: 10.1038/s41556-022-01072-x
Biologically informed deep learning to query gene programs in single-cell atlases
Abstract
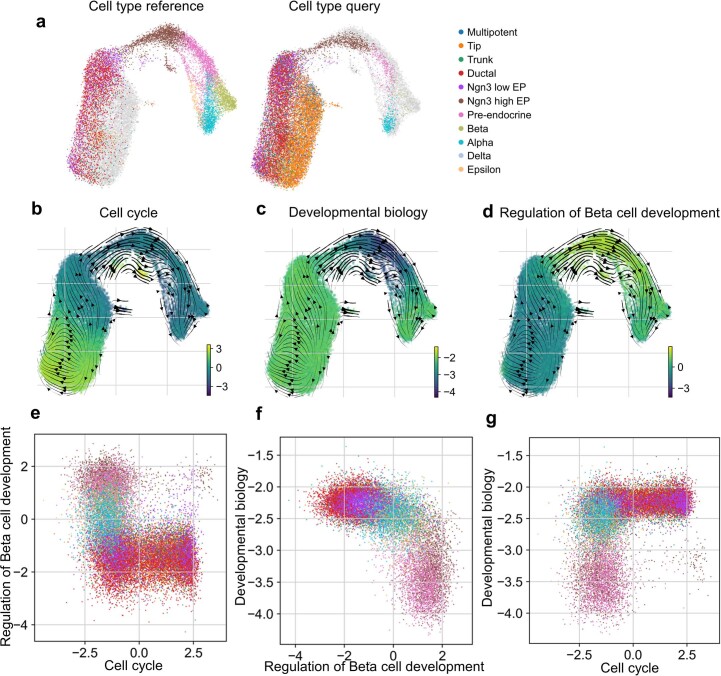
The increasing availability of large-scale single-cell atlases has enabled the detailed description of cell states. In parallel, advances in deep learning allow rapid analysis of newly generated query datasets by mapping them into reference atlases. However, existing data transformations learned to map query data are not easily explainable using biologically known concepts such as genes or pathways. Here we propose expiMap, a biologically informed deep-learning architecture that enables single-cell reference mapping. ExpiMap learns to map cells into biologically understandable components representing known 'gene programs'. The activity of each cell for a gene program is learned while simultaneously refining them and learning de novo programs. We show that expiMap compares favourably to existing methods while bringing an additional layer of interpretability to integrative single-cell analysis. Furthermore, we demonstrate its applicability to analyse single-cell perturbation responses in different tissues and species and resolve responses of patients who have coronavirus disease 2019 to different treatments across cell types.
© 2023. The Author(s).
Conflict of interest statement
F.J.T. consults for Immunai Inc., Singularity Bio B.V., CytoReason Ltd and Omniscope Ltd, and has ownership interest in Dermagnostix GmbH and Cellarity. The other authors declare no competing interests.
Figures















References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
