

Integrative multi-omics networks identify PKCδ and DNA-PK as master kinases of glioblastoma subtypes and guide targeted cancer therapy
- PMID: 36732634
- PMCID: PMC9970878
- DOI: 10.1038/s43018-022-00510-x
Integrative multi-omics networks identify PKCδ and DNA-PK as master kinases of glioblastoma subtypes and guide targeted cancer therapy
Abstract
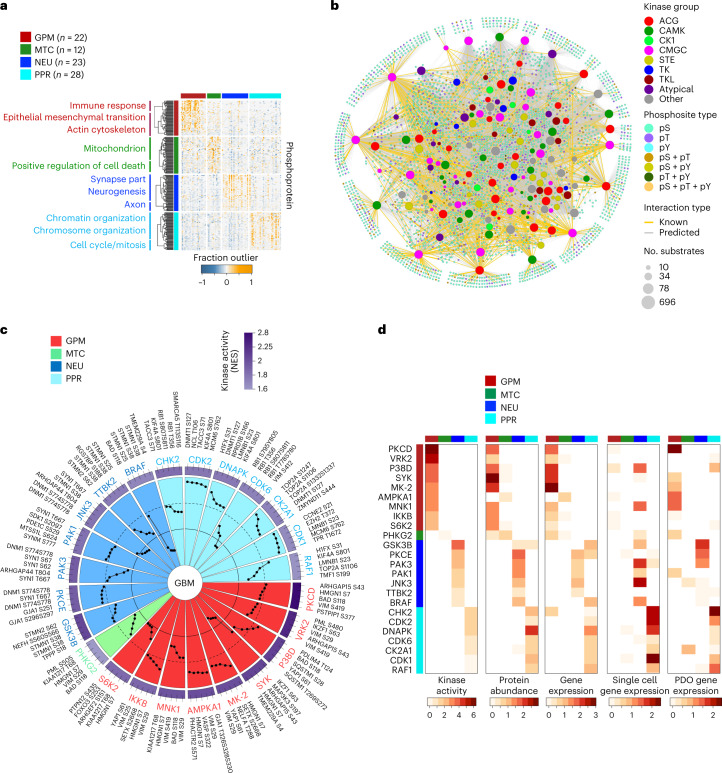
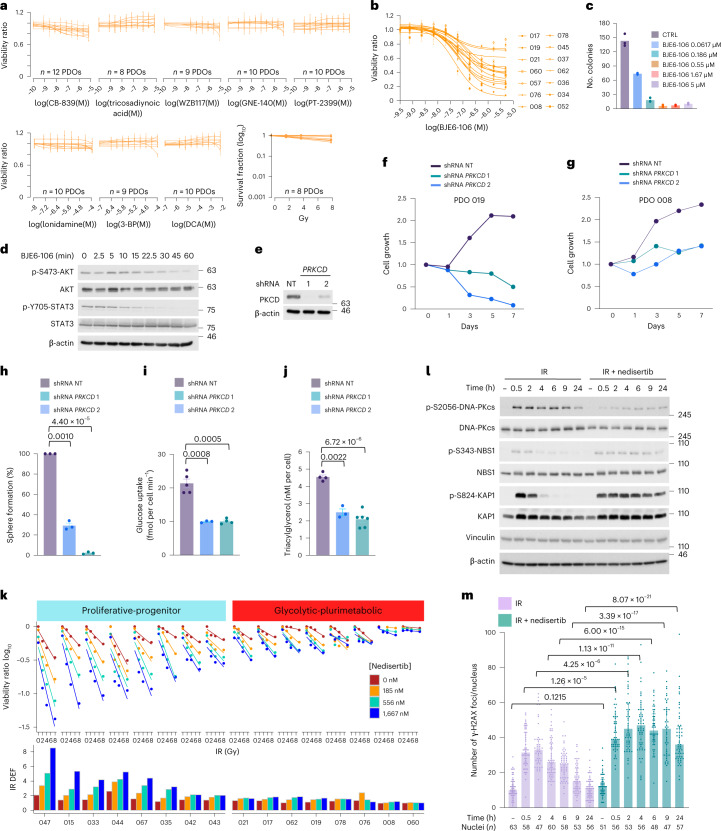
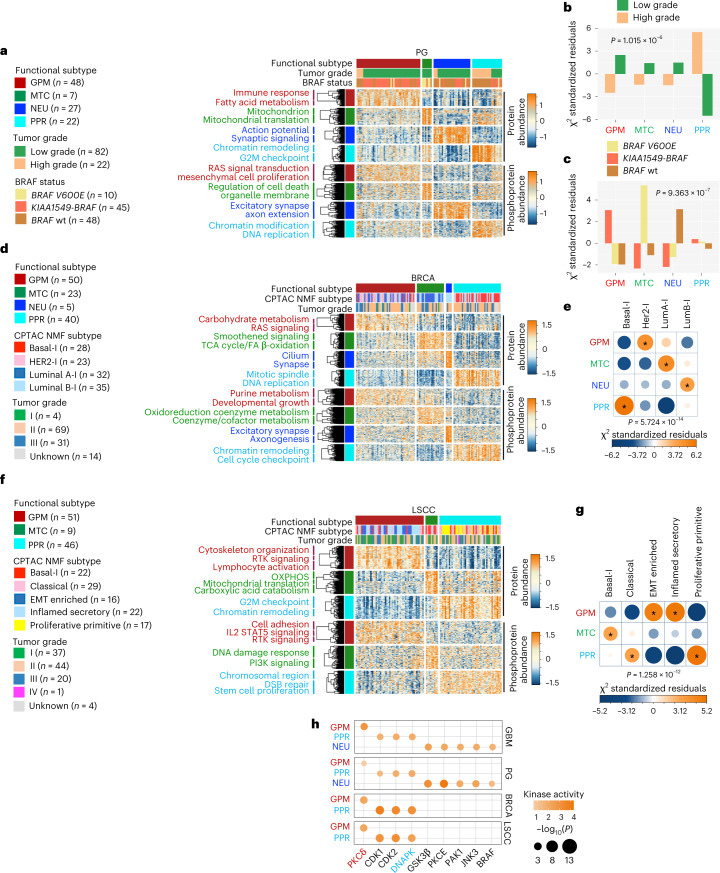
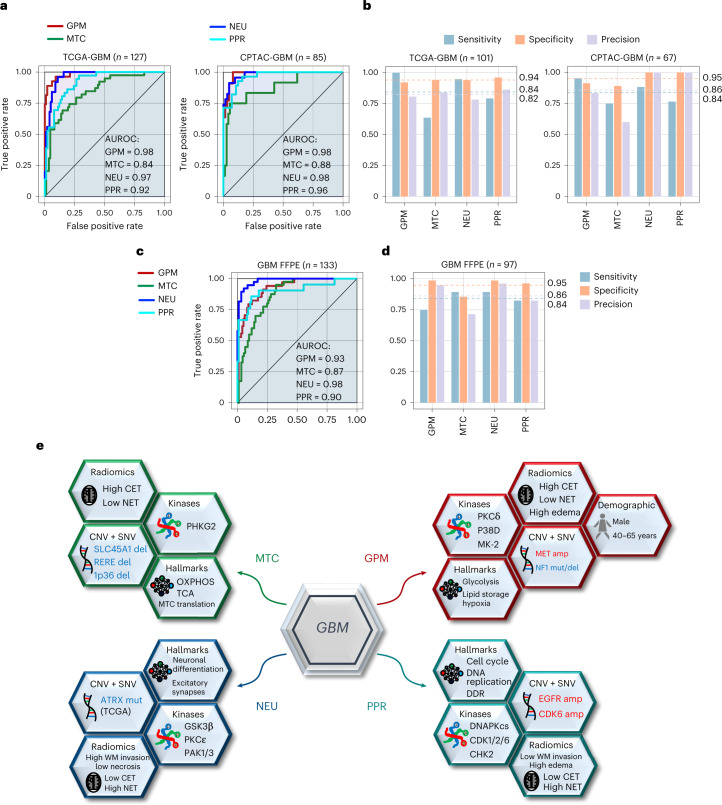
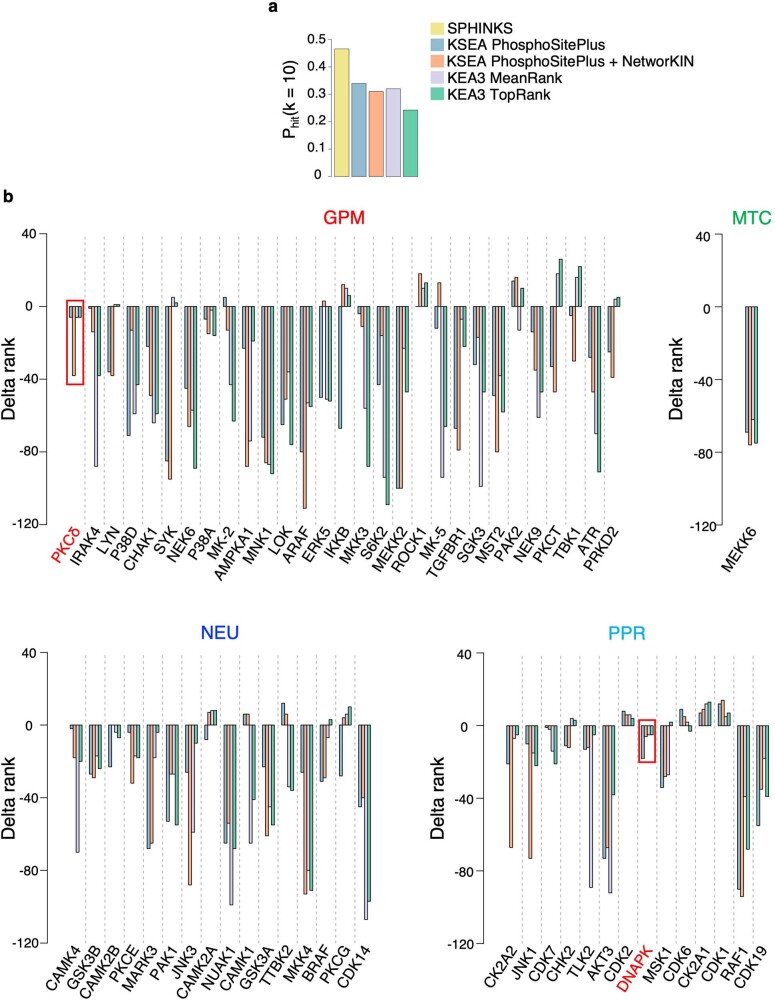
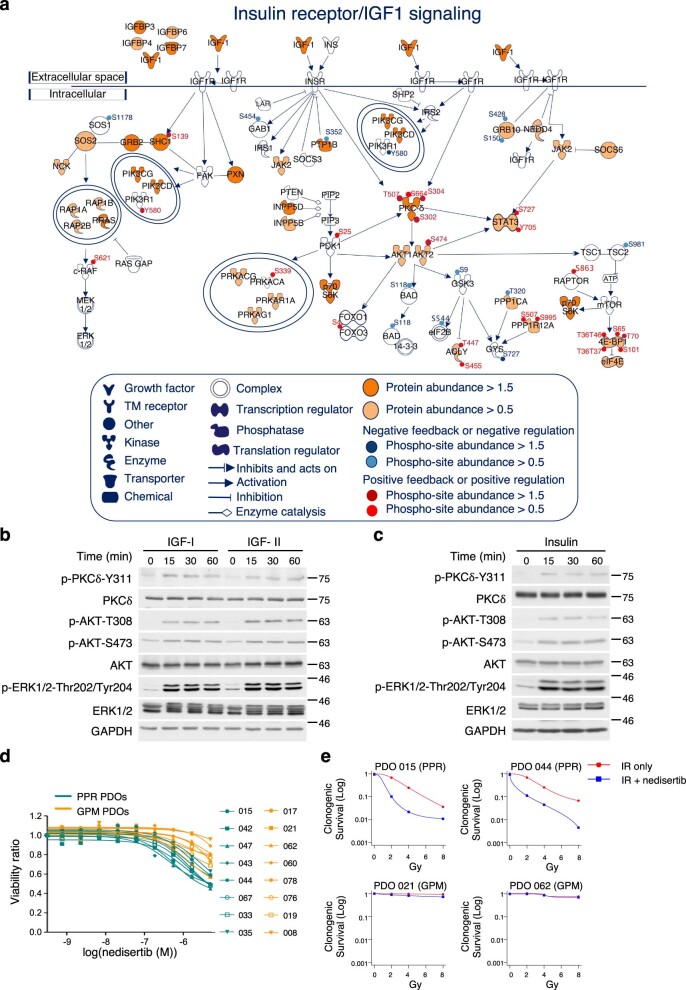
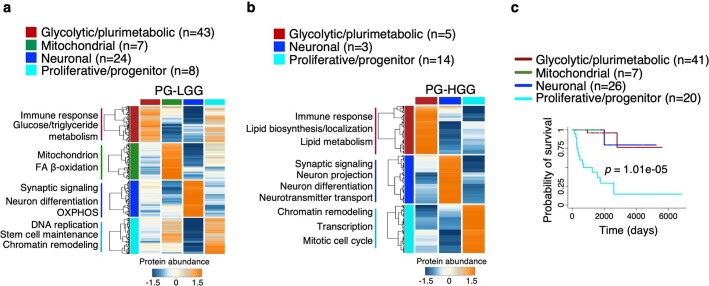
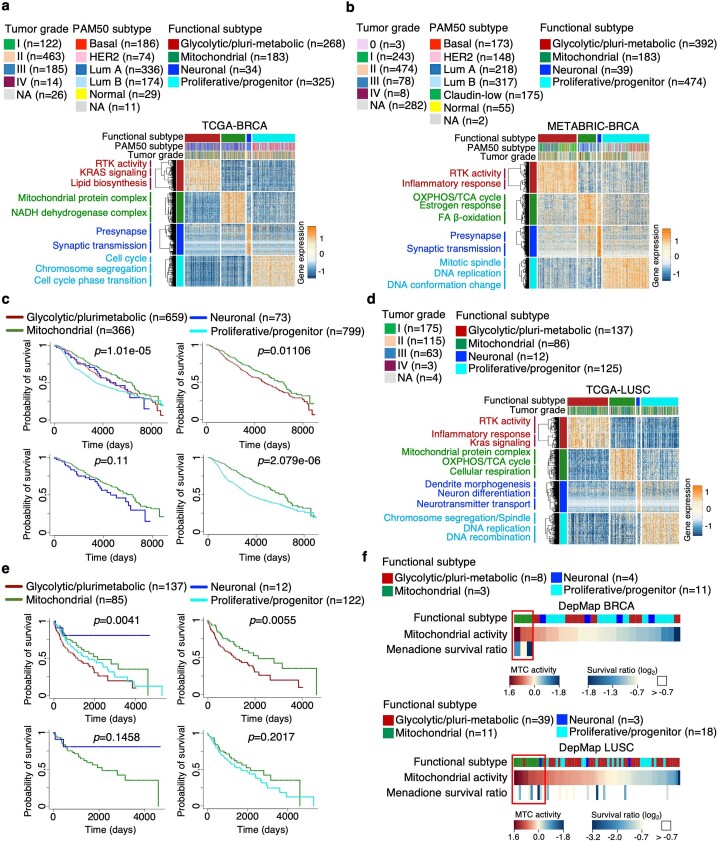
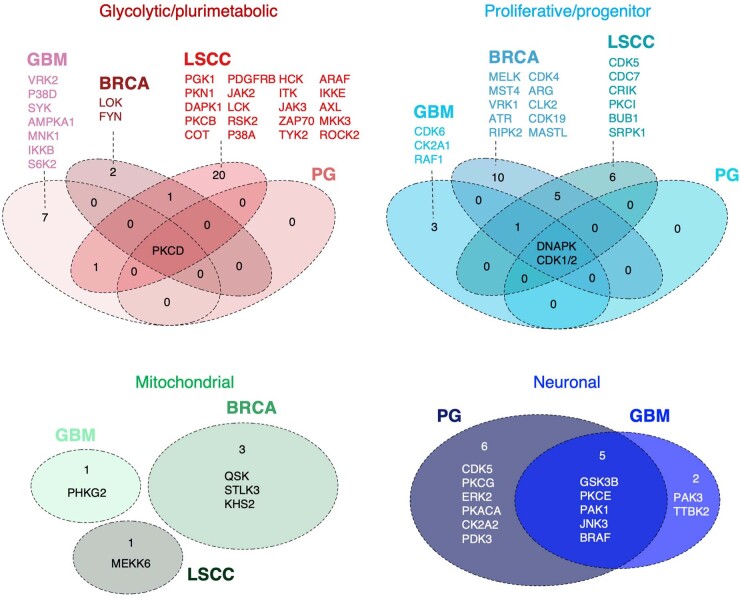
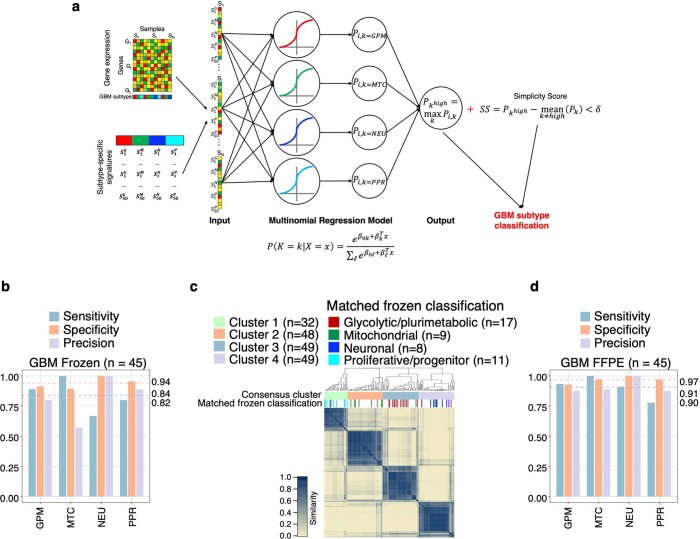
Despite producing a panoply of potential cancer-specific targets, the proteogenomic characterization of human tumors has yet to demonstrate value for precision cancer medicine. Integrative multi-omics using a machine-learning network identified master kinases responsible for effecting phenotypic hallmarks of functional glioblastoma subtypes. In subtype-matched patient-derived models, we validated PKCδ and DNA-PK as master kinases of glycolytic/plurimetabolic and proliferative/progenitor subtypes, respectively, and qualified the kinases as potent and actionable glioblastoma subtype-specific therapeutic targets. Glioblastoma subtypes were associated with clinical and radiomics features, orthogonally validated by proteomics, phospho-proteomics, metabolomics, lipidomics and acetylomics analyses, and recapitulated in pediatric glioma, breast and lung squamous cell carcinoma, including subtype specificity of PKCδ and DNA-PK activity. We developed a probabilistic classification tool that performs optimally with RNA from frozen and paraffin-embedded tissues, which can be used to evaluate the association of therapeutic response with glioblastoma subtypes and to inform patient selection in prospective clinical trials.
© 2023. The Author(s).
Conflict of interest statement
A.L. and A.I. are inventors of a biomarker technology that has been licensed to QIAGEN. A.I. received sponsored research funding from AstraZeneca and Taiho Pharmaceutical and has served as a paid consultant/advisor to AIMEDBIO. A.L. received sponsored research funding from Celgene. A.L. and A.I. are inventors of a patent application based on this work. All other authors declare no competing interests.
Figures
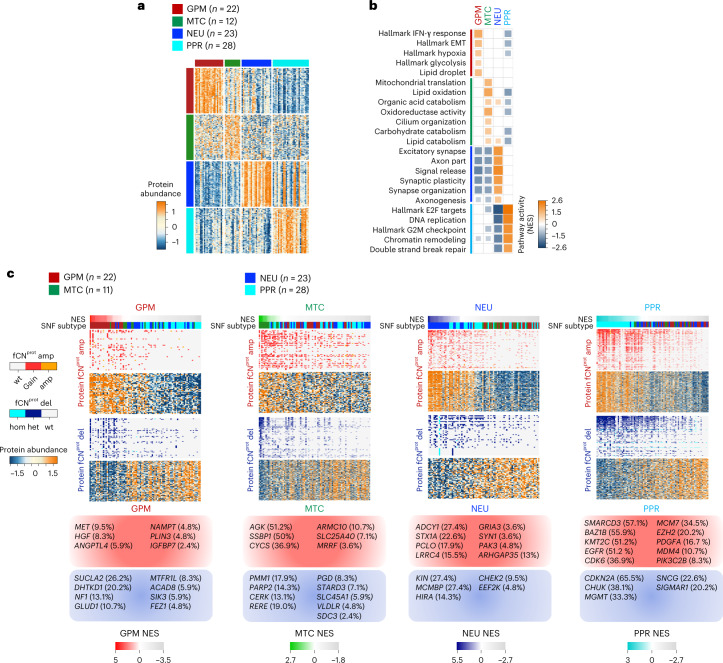
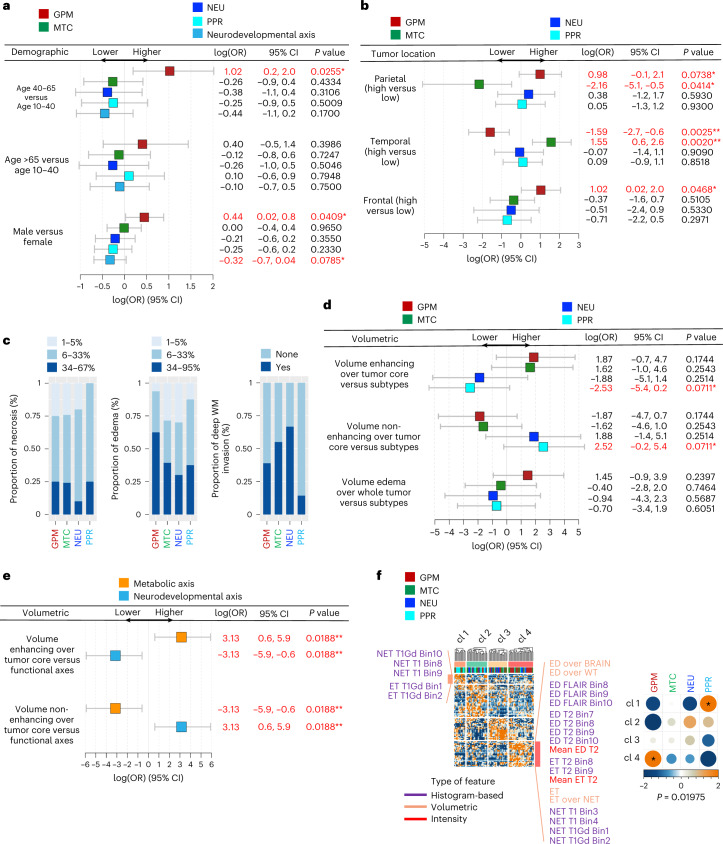
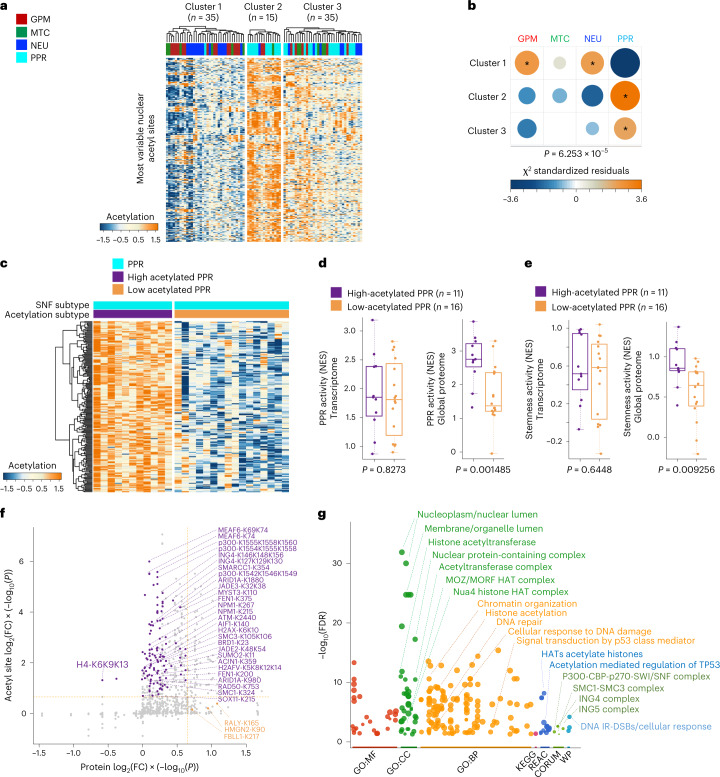
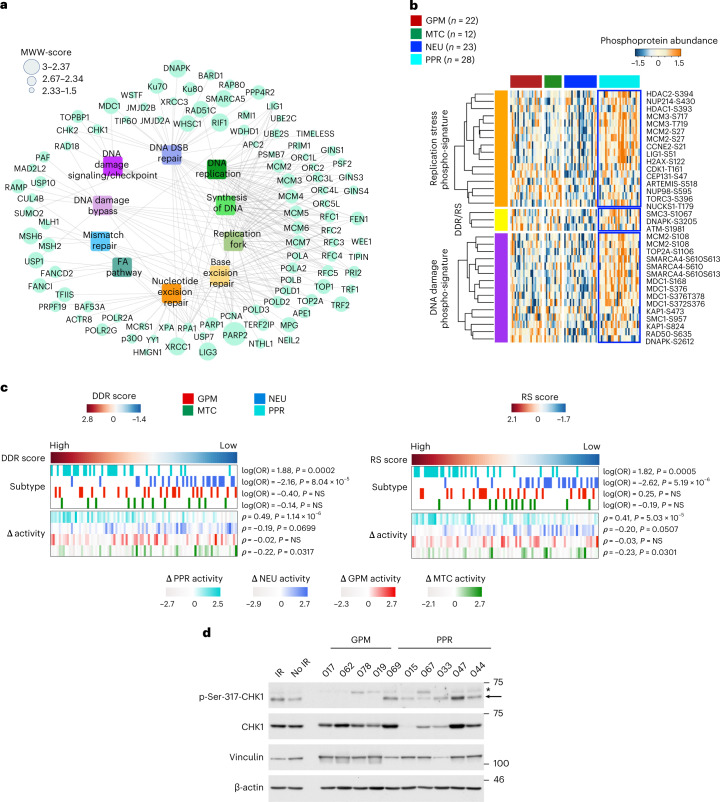
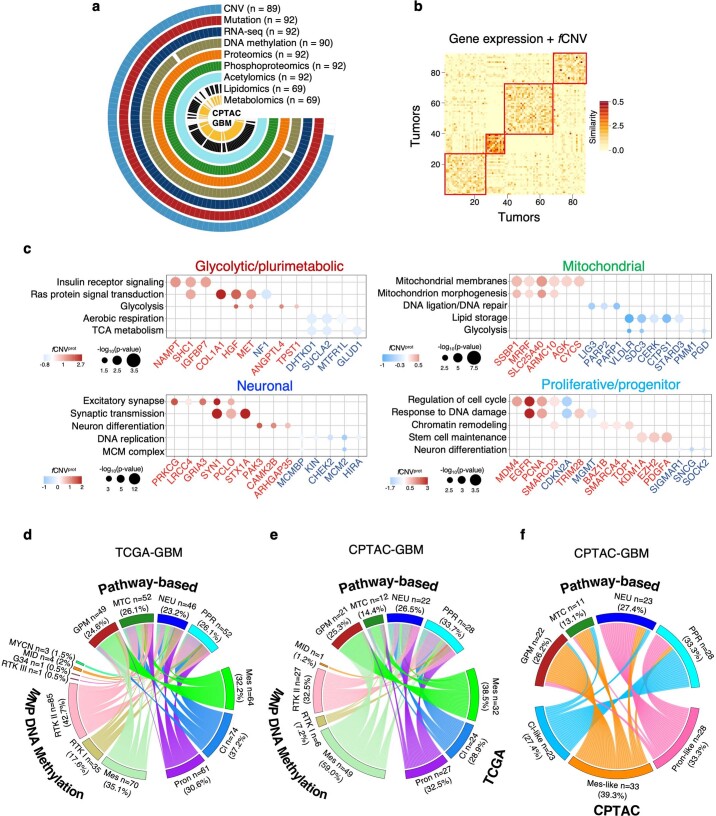
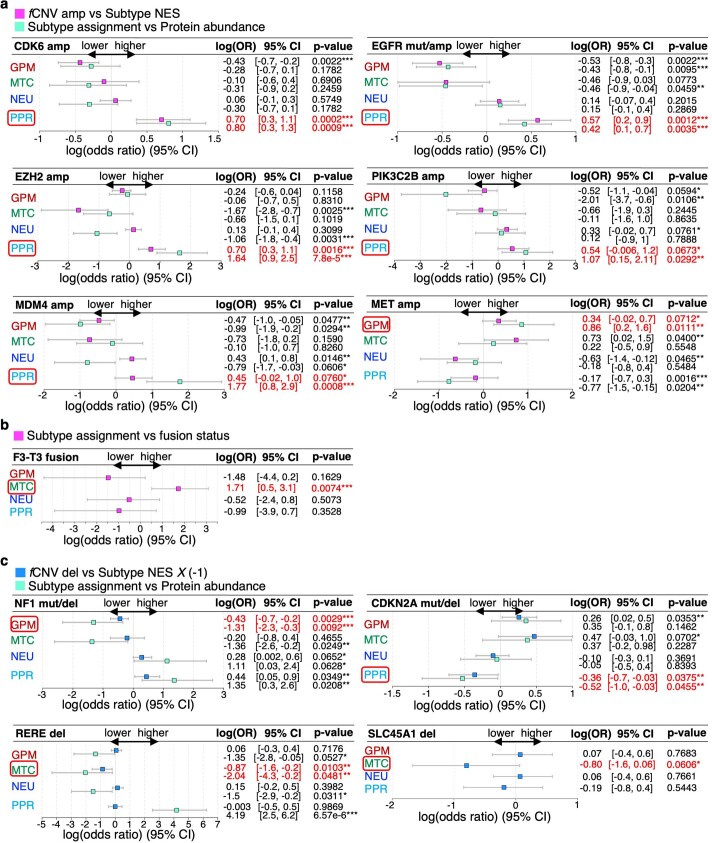
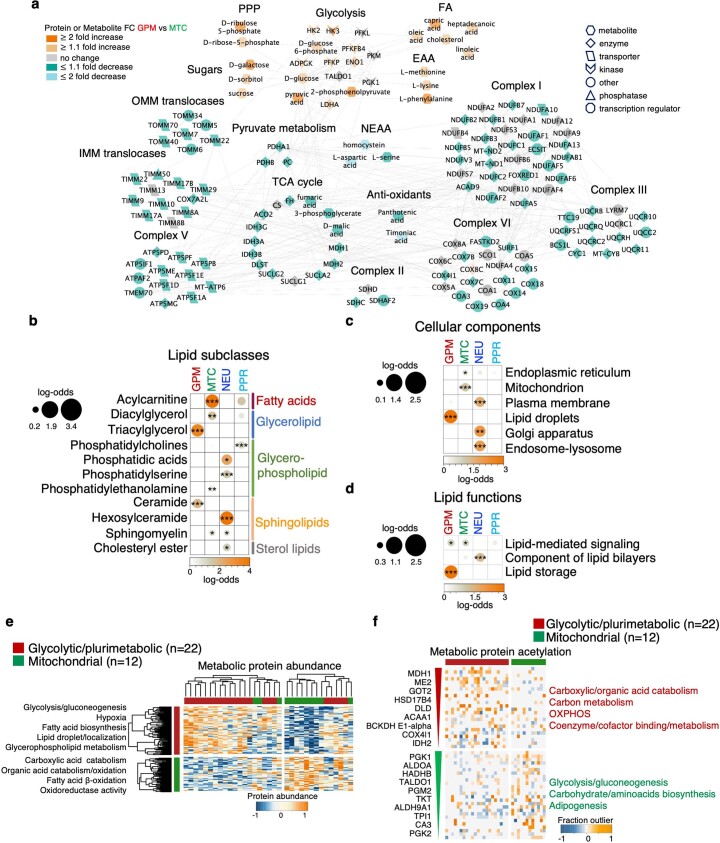
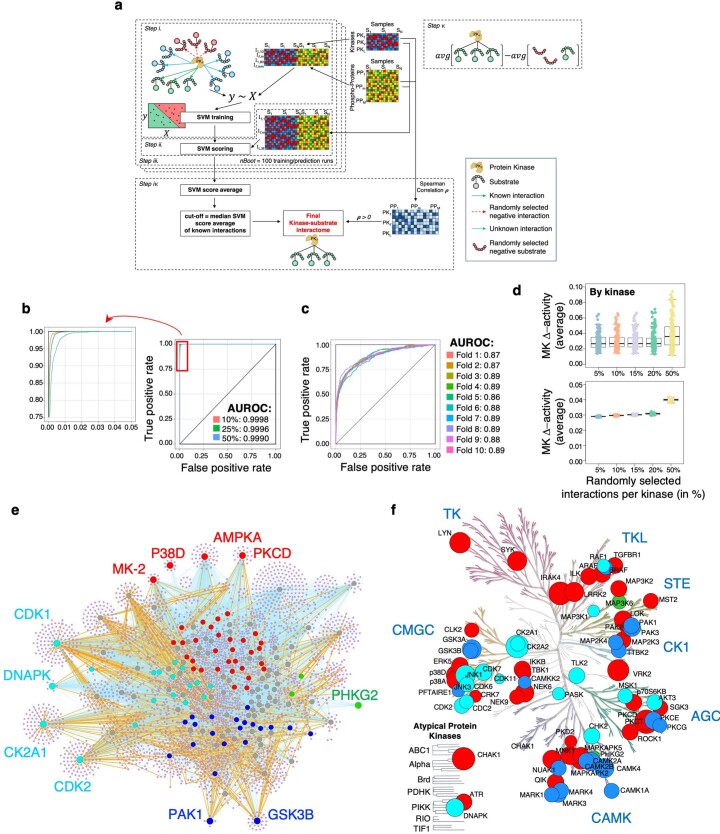
Comment in
-
Master kinases extracted from proteogenomic networks in glioblastoma drive cancer subtypes.Nat Cancer. 2023 Feb;4(2):159-160. doi: 10.1038/s43018-022-00511-w. Nat Cancer. 2023. PMID: 36732637 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R35CA253183/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- U54CA193313/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01CA268592/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01CA190891/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01CA239698/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
LinkOut - more resources
Full Text Sources
