

Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer's disease
- PMID: 36732642
- PMCID: PMC11332722
- DOI: 10.1038/s41593-022-01251-x
Integrative in situ mapping of single-cell transcriptional states and tissue histopathology in a mouse model of Alzheimer's disease
Abstract
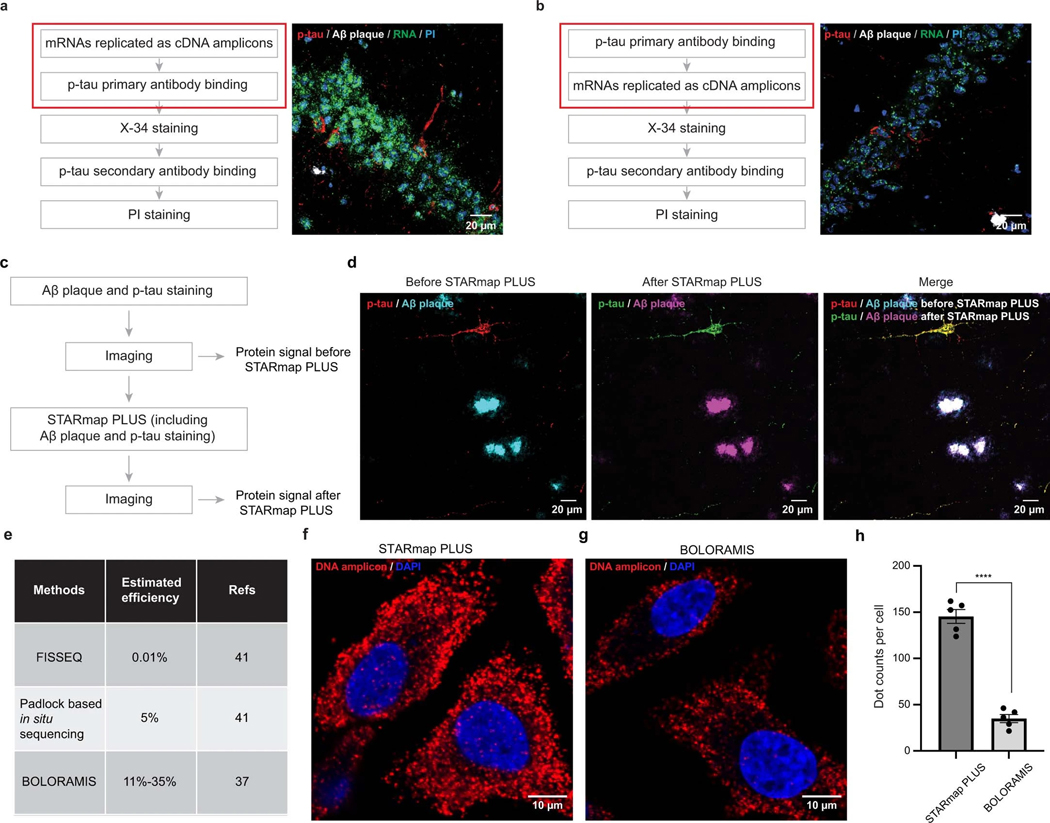
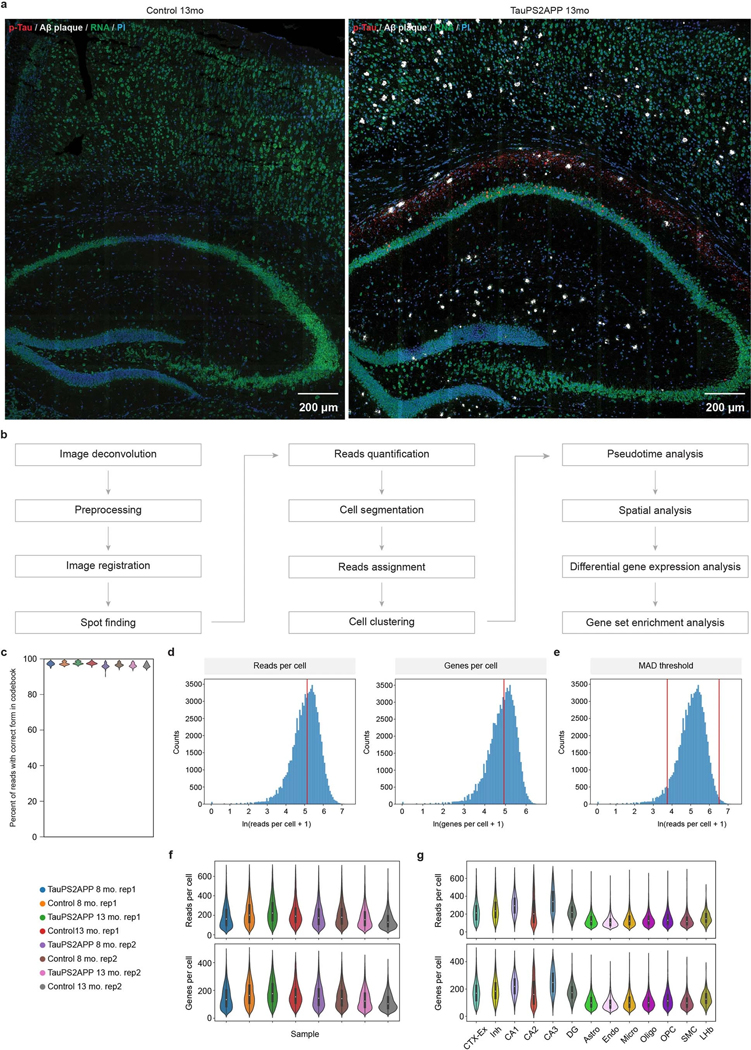
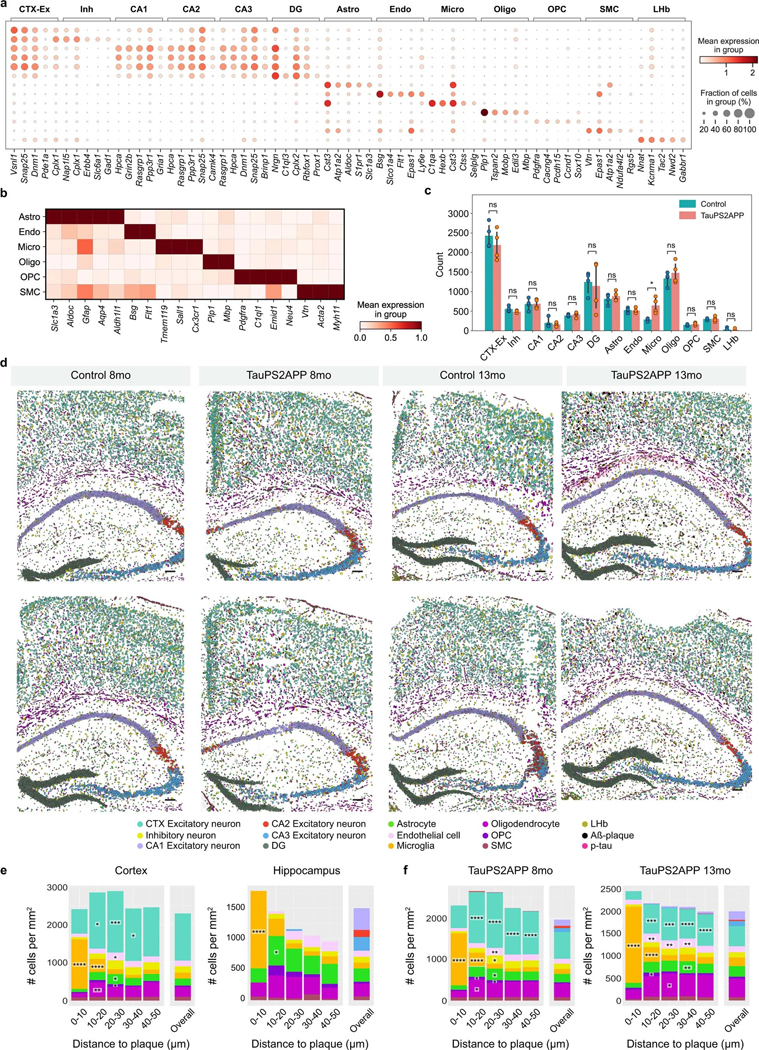
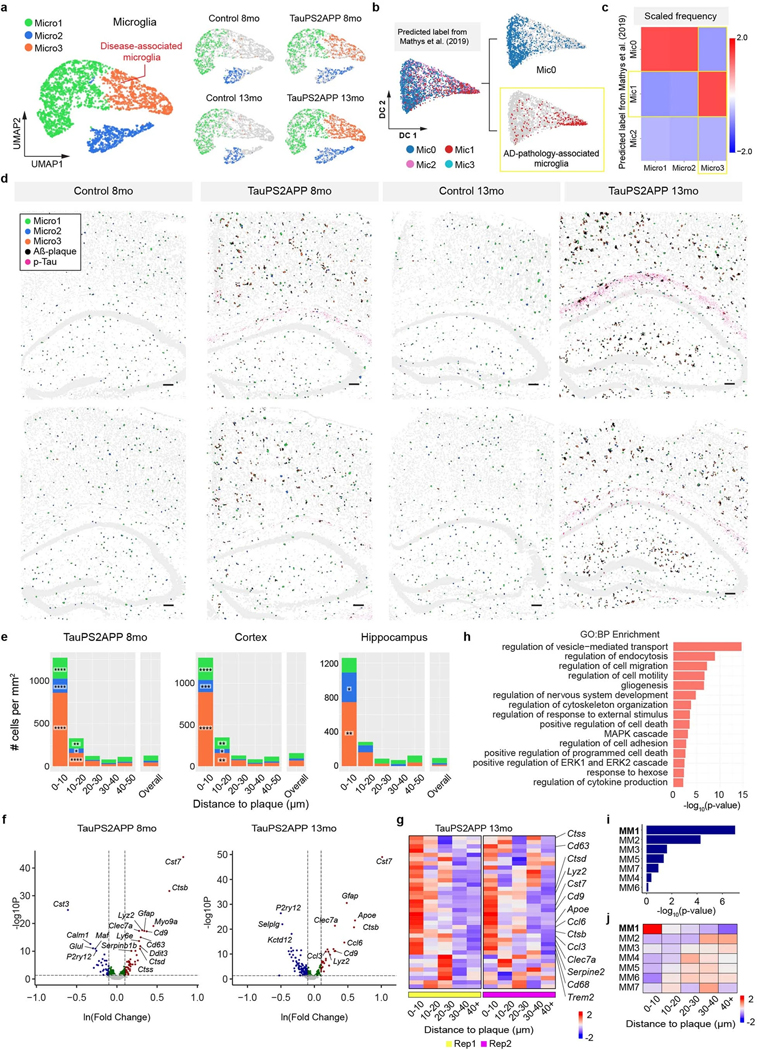
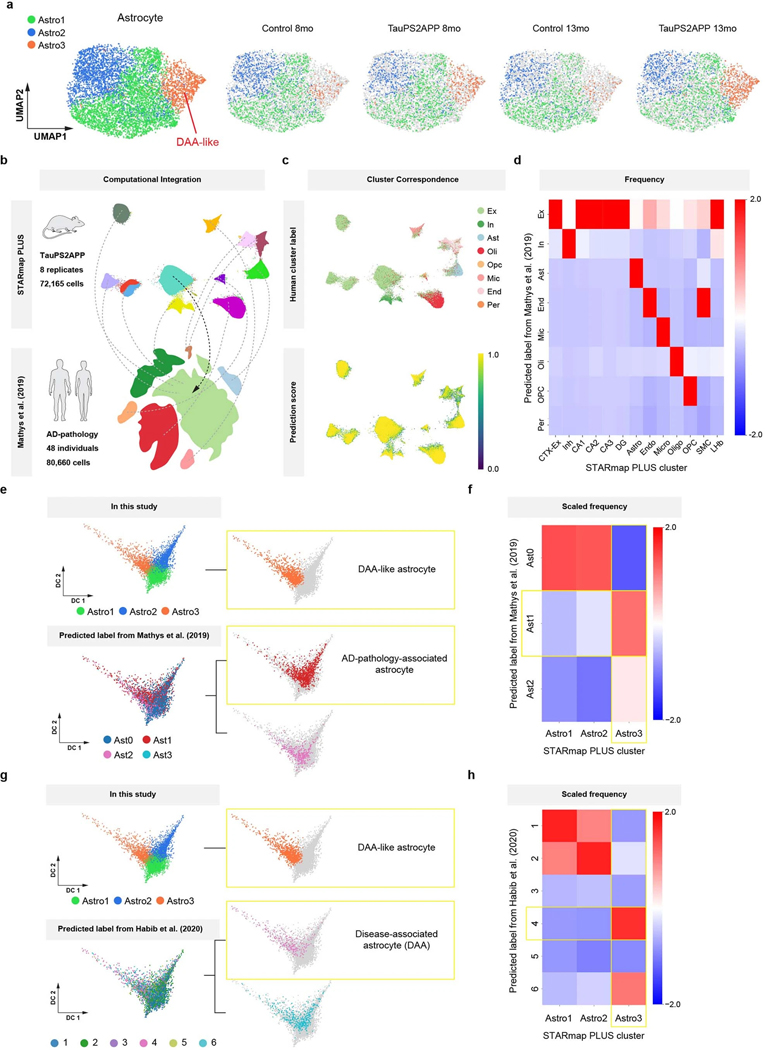
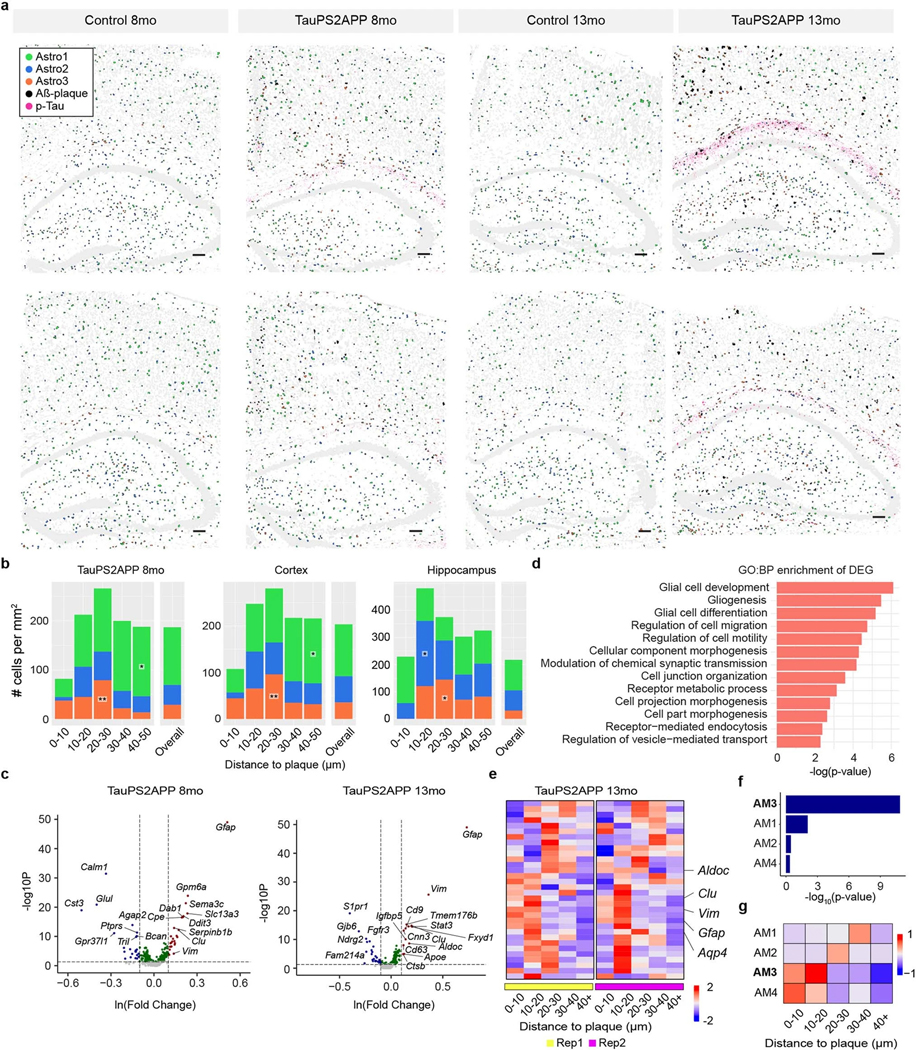
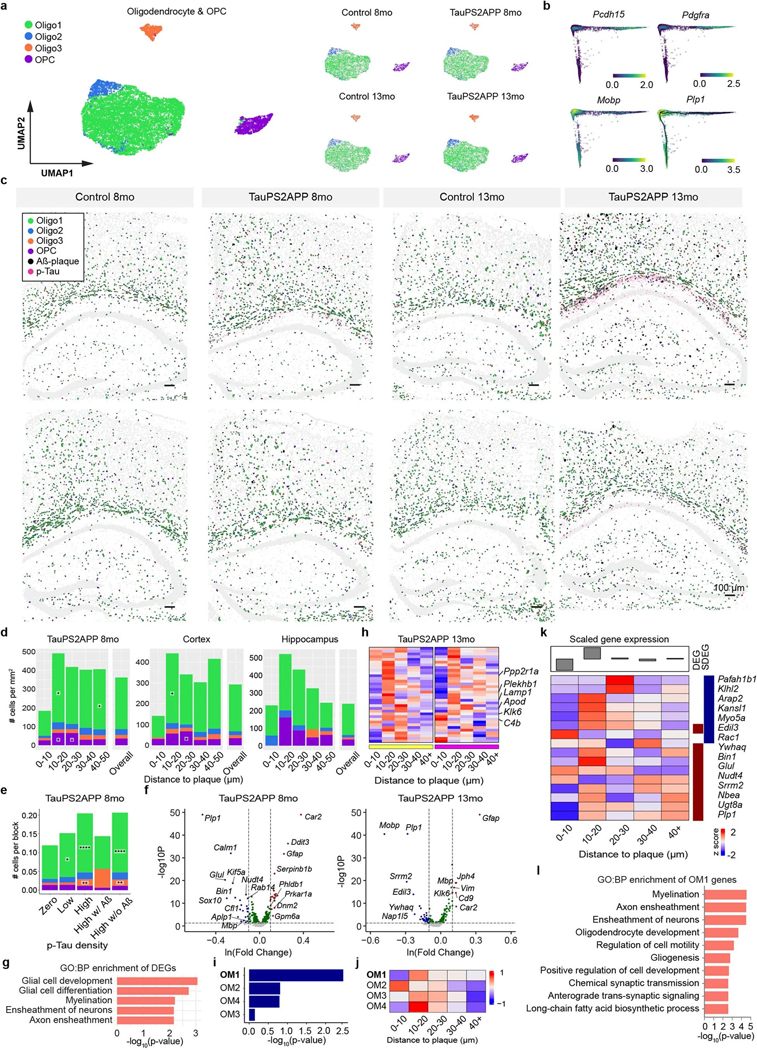
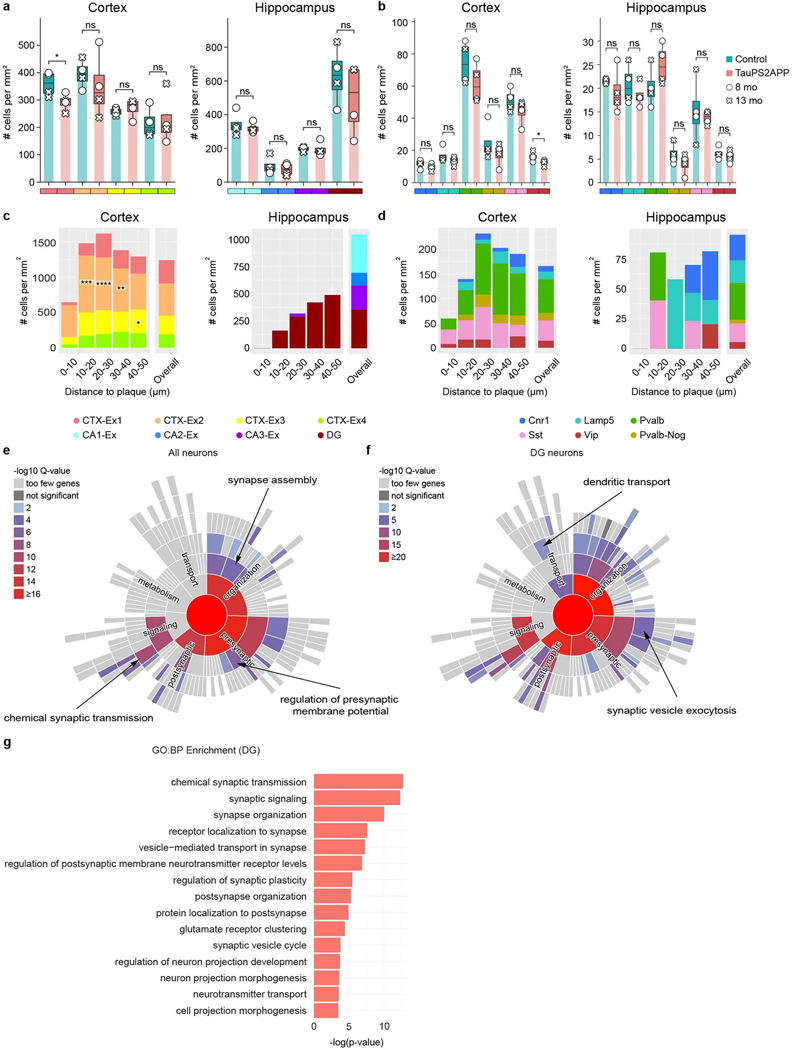
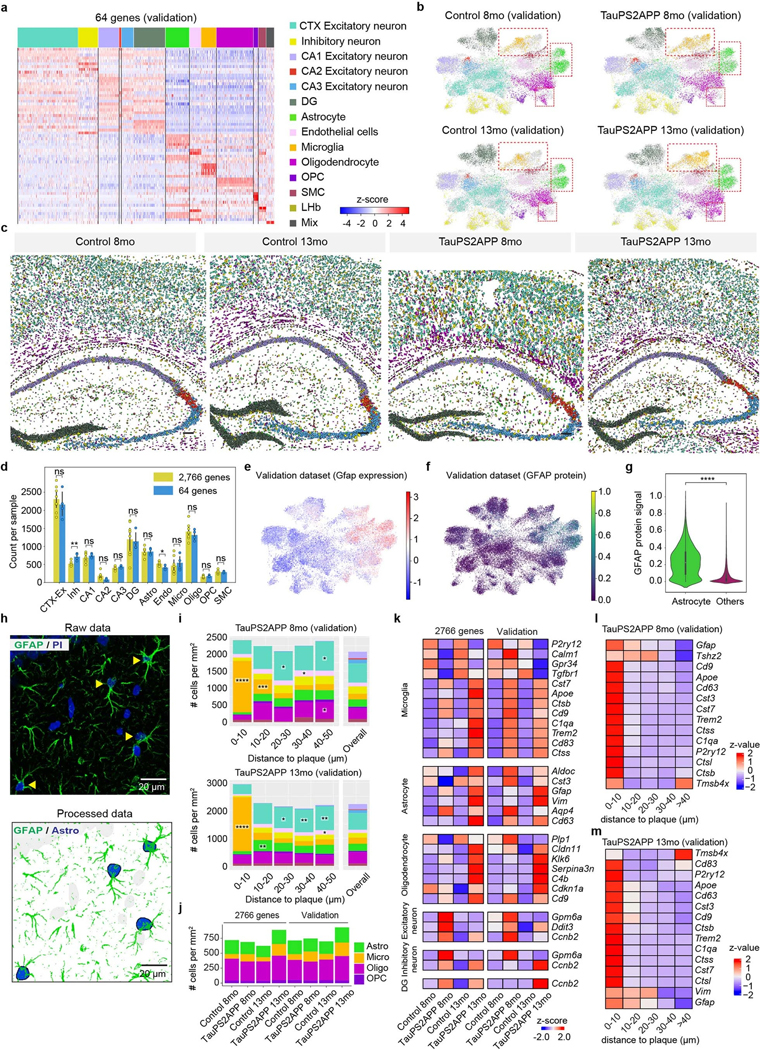
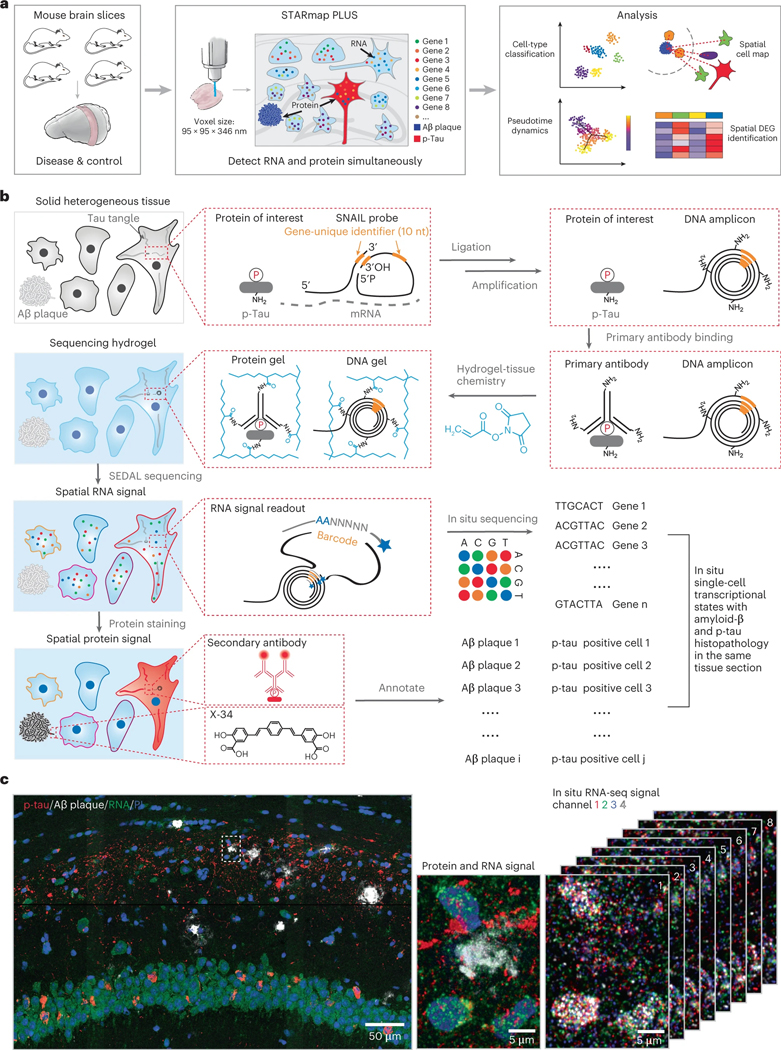
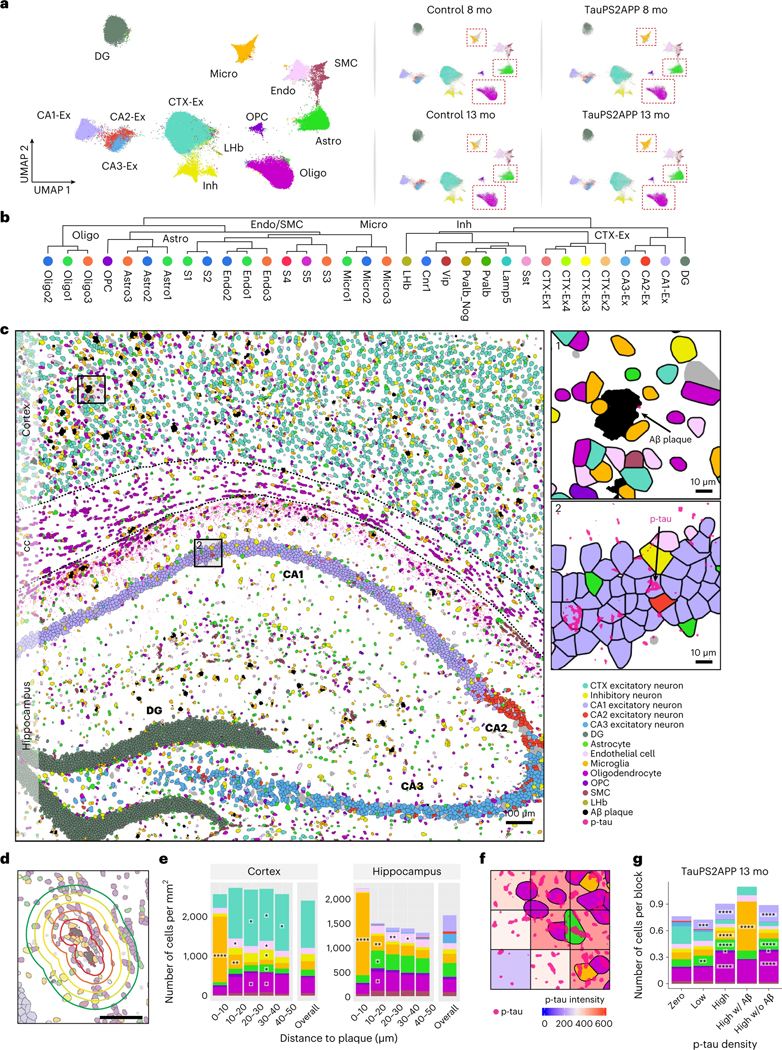
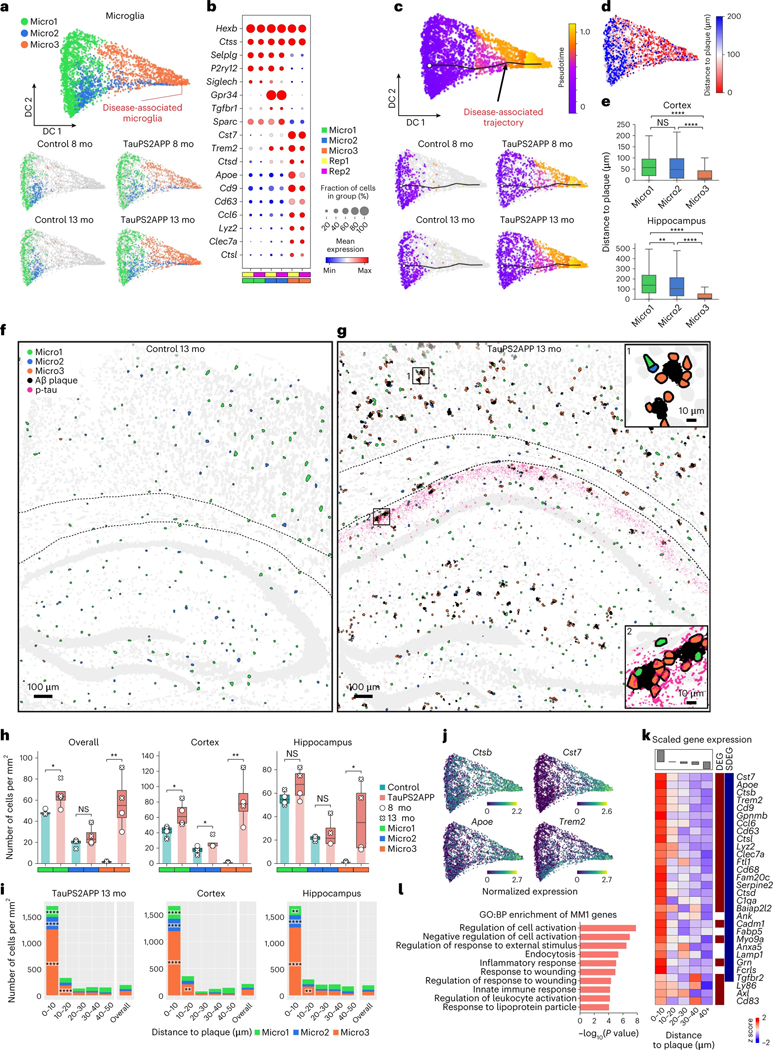
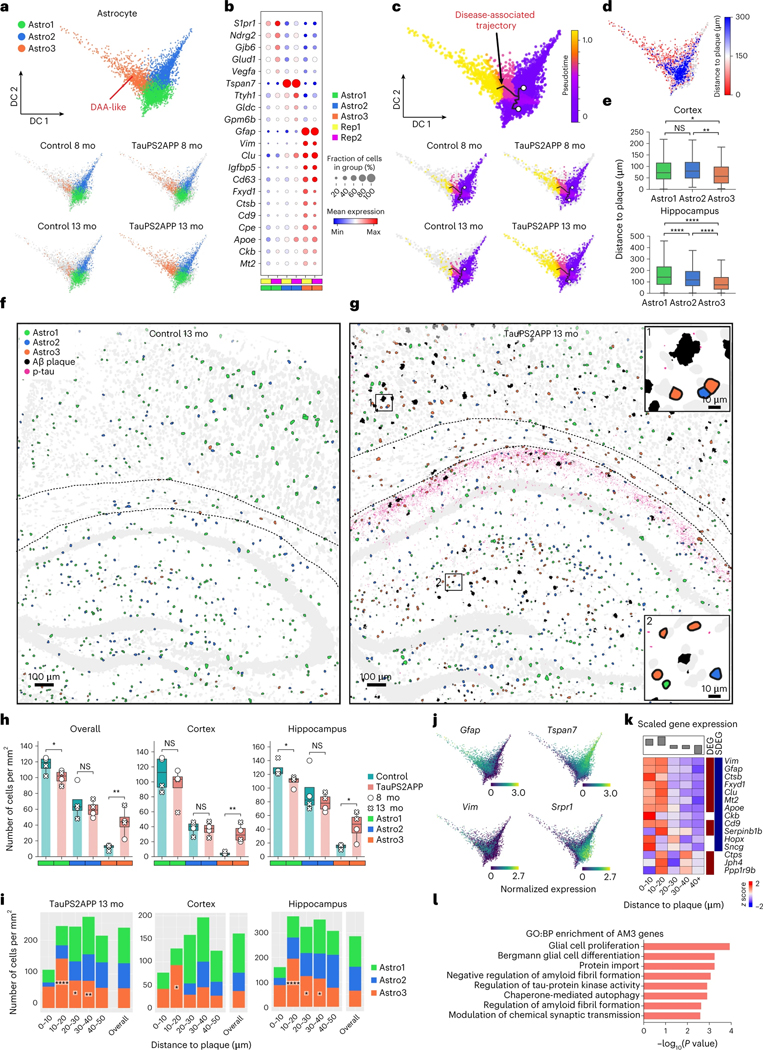
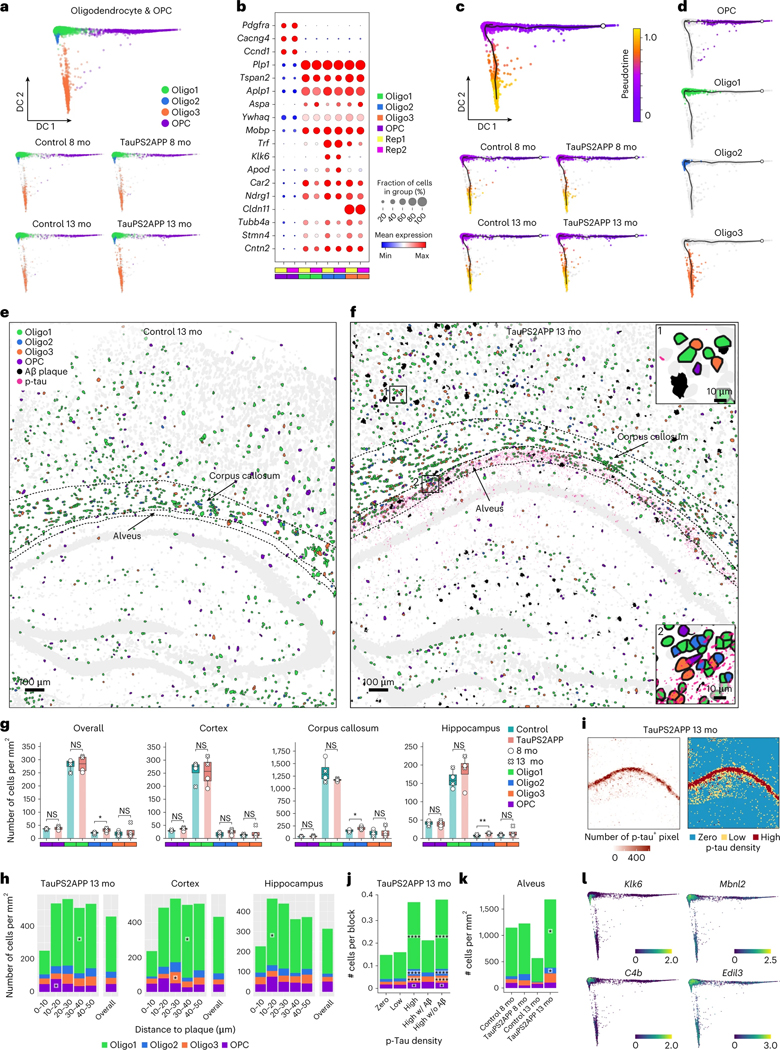
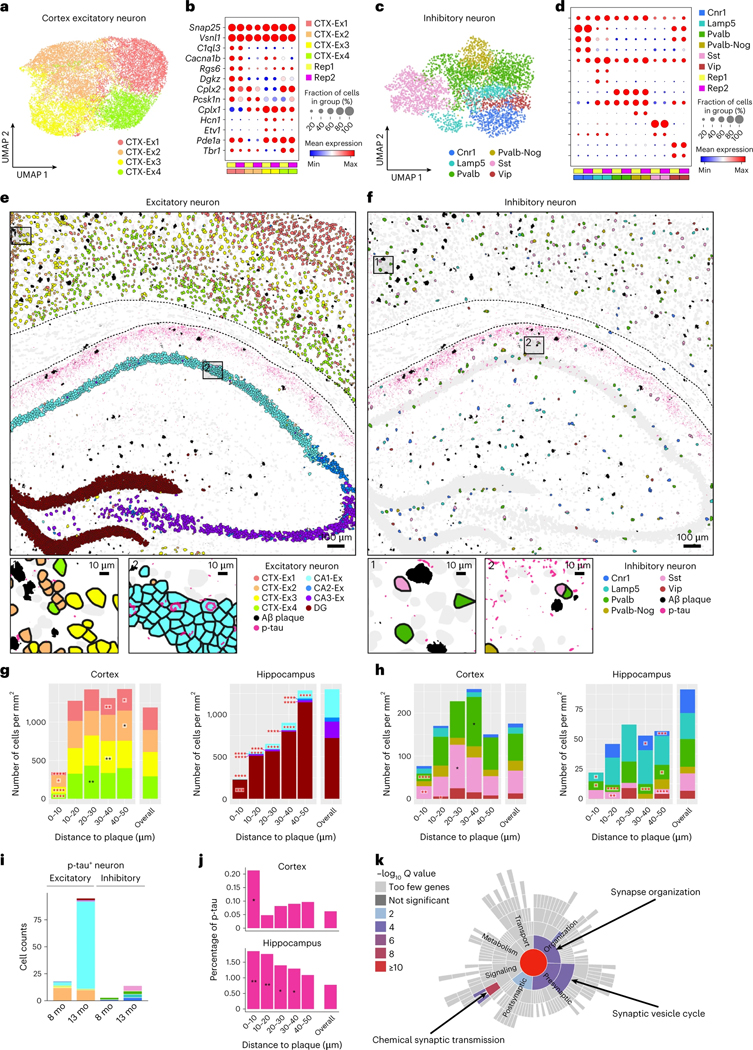
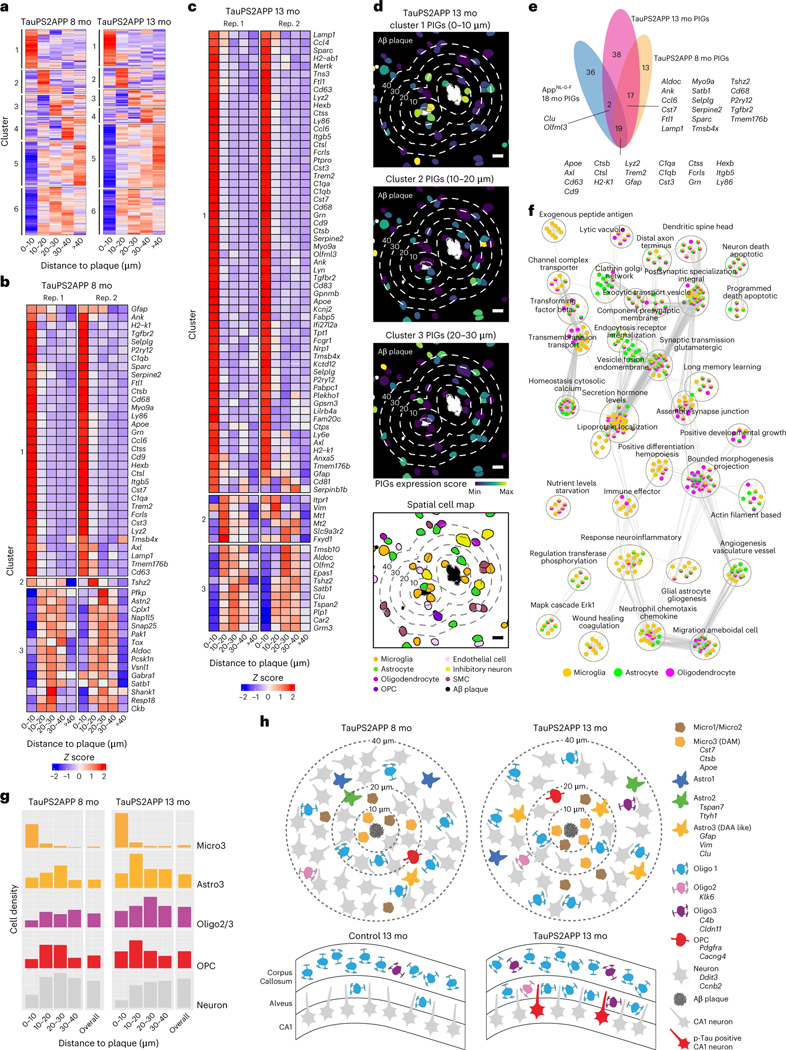
Complex diseases are characterized by spatiotemporal cellular and molecular changes that may be difficult to comprehensively capture. However, understanding the spatiotemporal dynamics underlying pathology can shed light on disease mechanisms and progression. Here we introduce STARmap PLUS, a method that combines high-resolution spatial transcriptomics with protein detection in the same tissue section. As proof of principle, we analyze brain tissues of a mouse model of Alzheimer's disease at 8 and 13 months of age. Our approach provides a comprehensive cellular map of disease progression. It reveals a core-shell structure where disease-associated microglia (DAM) closely contact amyloid-β plaques, whereas disease-associated astrocyte-like (DAA-like) cells and oligodendrocyte precursor cells (OPCs) are enriched in the outer shells surrounding the plaque-DAM complex. Hyperphosphorylated tau emerges mainly in excitatory neurons in the CA1 region and correlates with the local enrichment of oligodendrocyte subtypes. The STARmap PLUS method bridges single-cell gene expression profiles with tissue histopathology at subcellular resolution, providing a tool to pinpoint the molecular and cellular changes underlying pathology.
© 2023. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
X.W., H.Z. and J.R. are inventors on pending patent applications related to STARmap PLUS. X.W. is a scientific cofounder of Stellaromics. M.S. is a scientific cofounder of Neumora and a former employee of Genentech. W.J.M., C. J. B. and S.-H.L. are present employees of Genentech. All the other authors declare no competing interests.
Figures

References
-
- Masters CL et al. Alzheimer’s disease. Nat. Rev. Dis. Prim. 1, 15056 (2015). - PubMed
-
- Braak H. & Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991). - PubMed
-
- Hardy J. & Selkoe DJ The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002). - PubMed
-
- Beach TG, Walker R. & McGeer EG Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia 2, 420–436 (1989). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
