

Familial Alzheimer's Disease-Related Mutations Differentially Alter Stability of Amyloid-Beta Aggregates
- PMID: 36734539
- PMCID: PMC9940190
- DOI: 10.1021/acs.jpclett.2c03729
Familial Alzheimer's Disease-Related Mutations Differentially Alter Stability of Amyloid-Beta Aggregates
Abstract
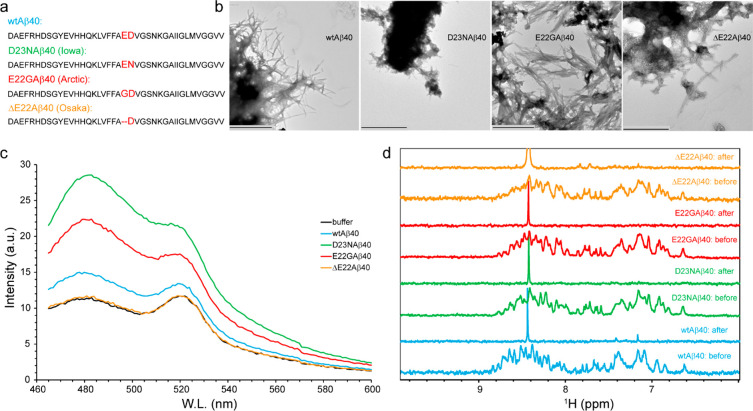
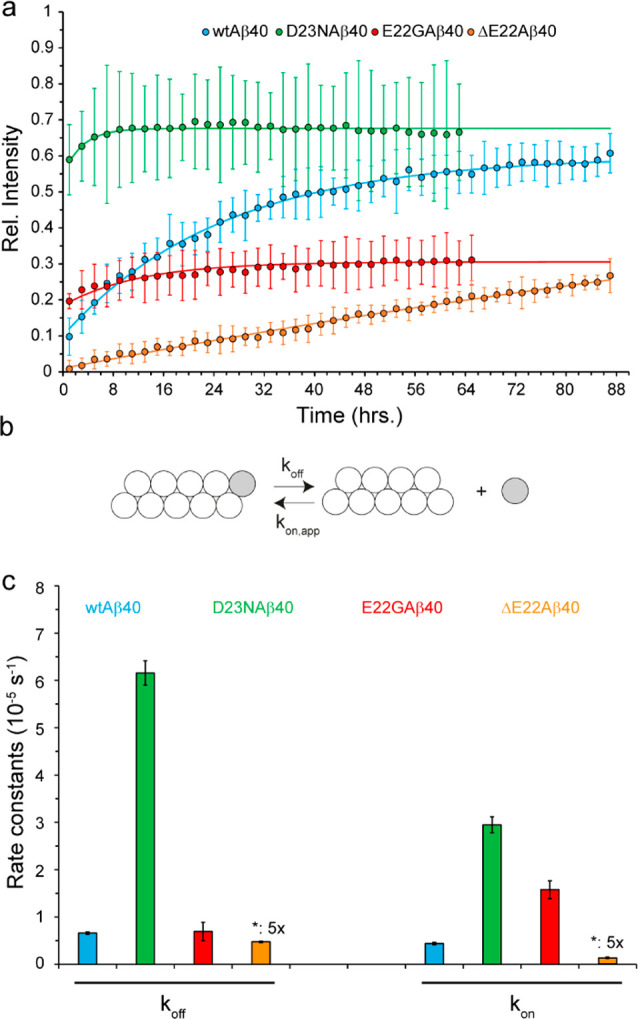
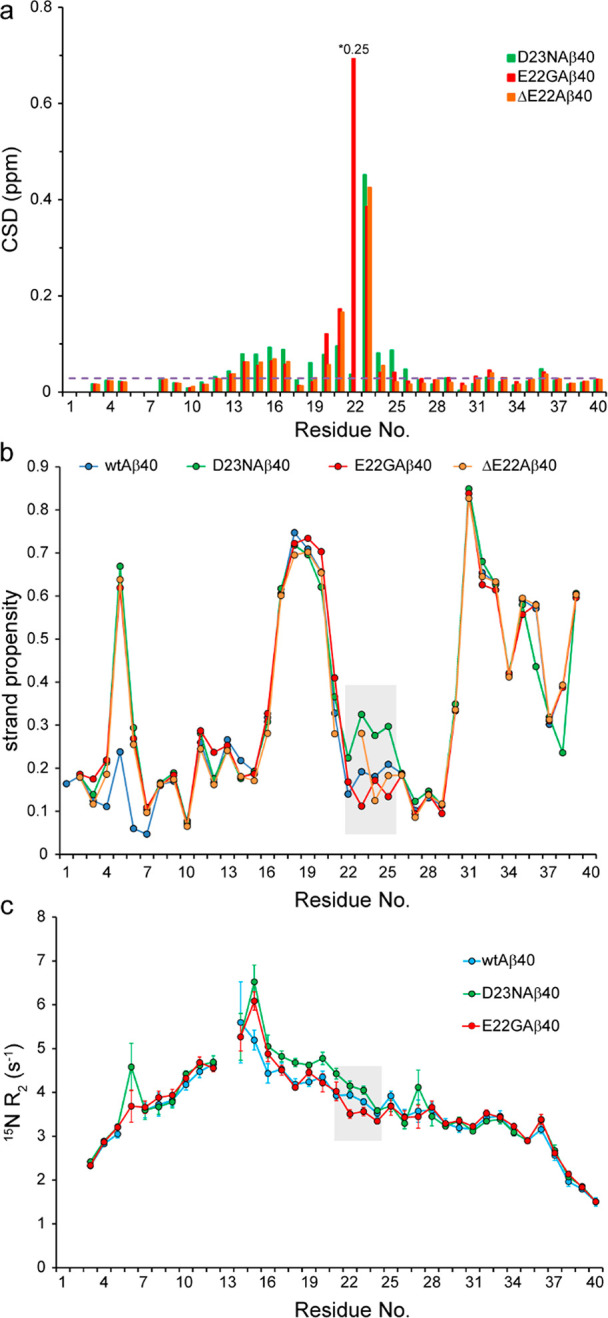
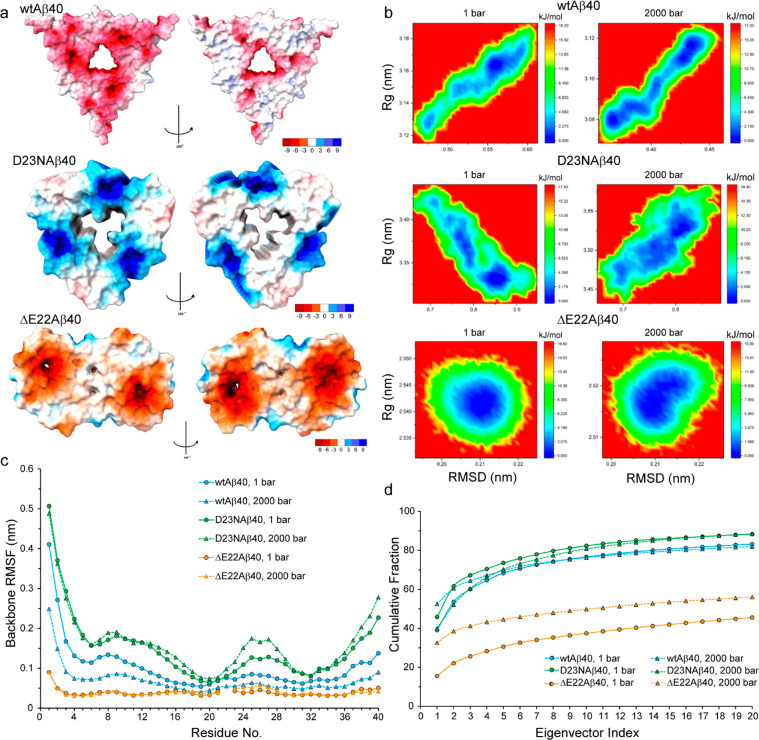
Amyloid-beta (Aβ) deposition as senile plaques is a pathological hallmark of Alzheimer's disease (AD). AD is characterized by a large level of heterogeneity in amyloid pathology, whose molecular origin is poorly understood. Here, we employ NMR spectroscopy and MD simulation at ambient and high pressures and investigate how AD-related mutations in Aβ peptide influence the stability of Aβ aggregates. The pressure-induced monomer dissociation from Aβ aggregates monitored by NMR demonstrated that the Iowa (D23N), Arctic (E22G), and Osaka (ΔE22) mutations altered the pressure stability of Aβ40 aggregates in distinct manners. While the NMR data of monomeric Aβ40 showed only small localized effects of mutations, the MD simulation of mutated Aβ fibrils revealed their distinct susceptibility to elevated pressure. Our data propose a structural basis for the distinct stability of various Aβ fibrils and highlights "stability" as a molecular property potentially contributing to the large heterogeneity of amyloid pathology in AD.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- World Health Organization. Dementia; https://www.who.int/en/news-room/fact-sheets/detail/dementia (accessed Jan. 21, 2023).
-
- Ryman D. C.; Acosta-Baena N.; Aisen P. S.; Bird T.; Danek A.; Fox N. C.; Goate A.; Frommelt P.; Ghetti B.; Langbaum J. B.; Lopera F.; Martins R.; Masters C. L.; Mayeux R. P.; McDade E.; Moreno S.; Reiman E. M.; Ringman J. M.; Salloway S.; Schofield P. R.; Sperling R.; Tariot P. N.; Xiong C.; Morris J. C.; Bateman R. J.; Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 2014, 83, 253–260. 10.1212/WNL.0000000000000596. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
