

Osteoarthritis: pathogenic signaling pathways and therapeutic targets
- PMID: 36737426
- PMCID: PMC9898571
- DOI: 10.1038/s41392-023-01330-w
Osteoarthritis: pathogenic signaling pathways and therapeutic targets
Abstract
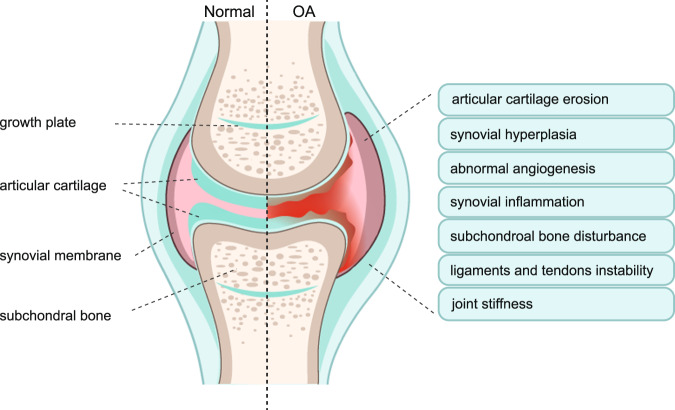
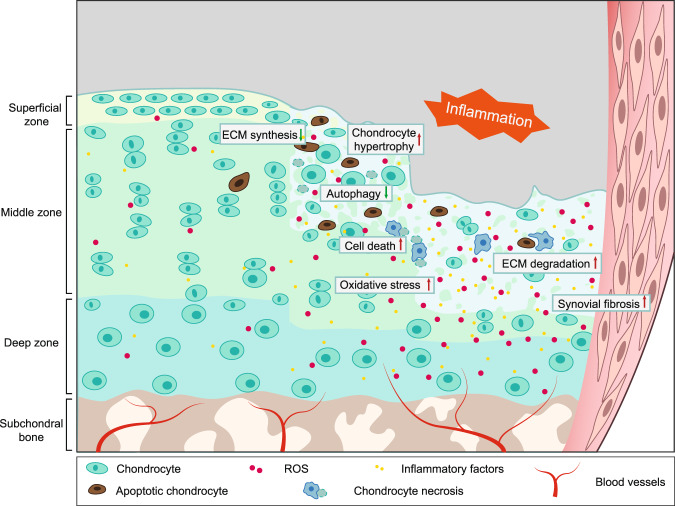
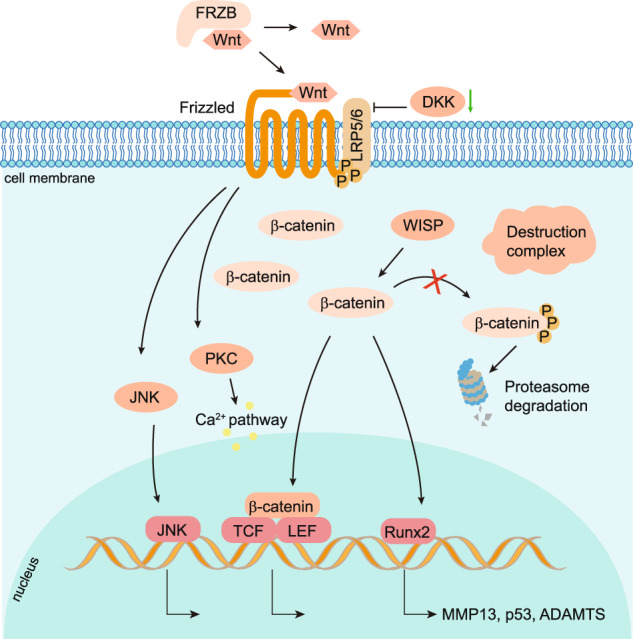
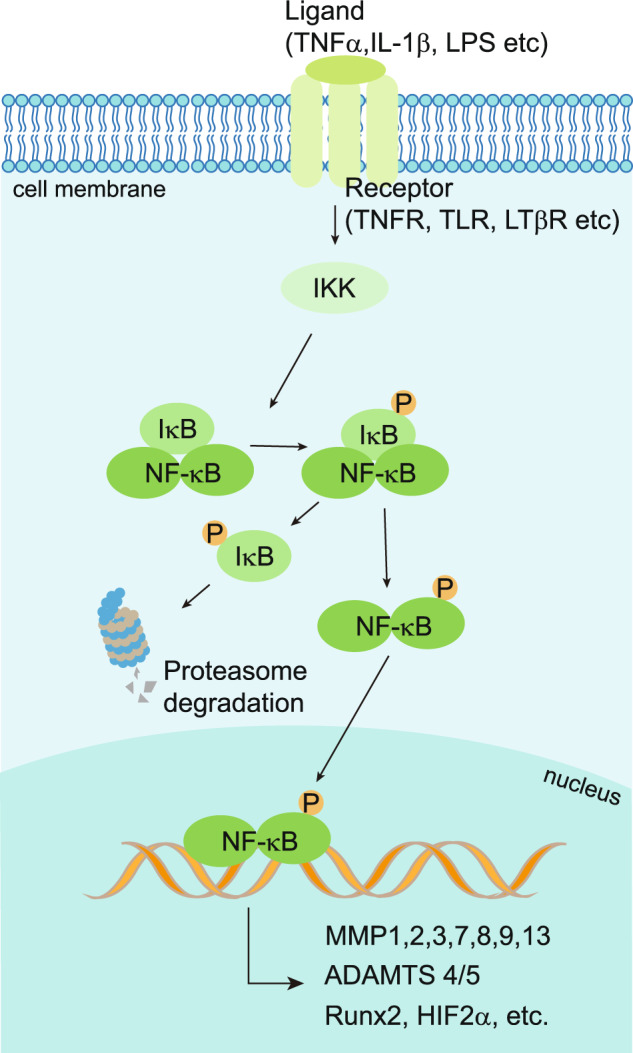
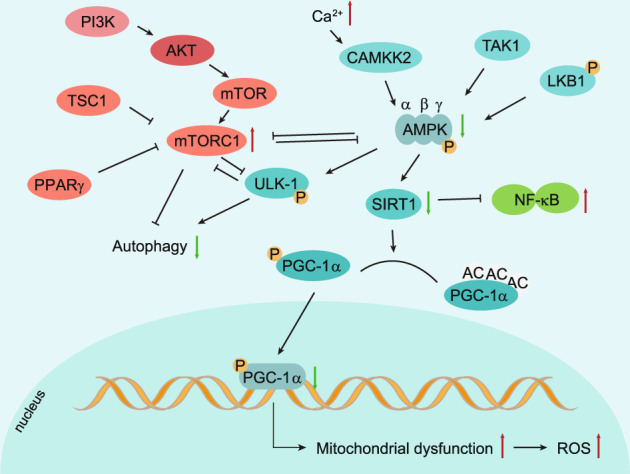
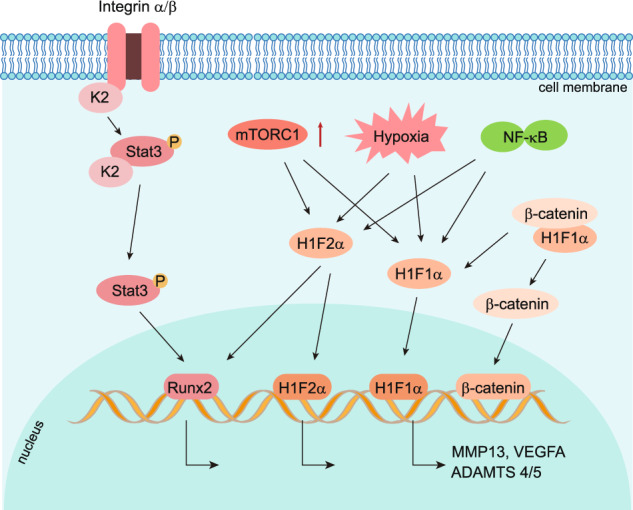
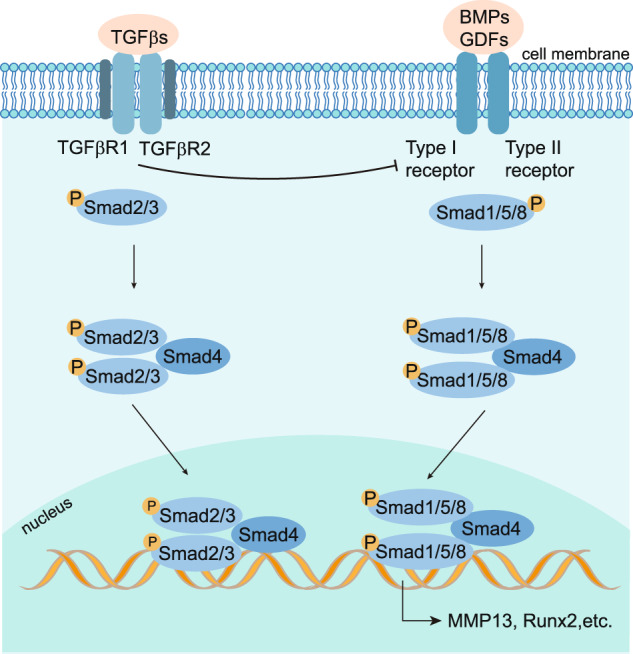
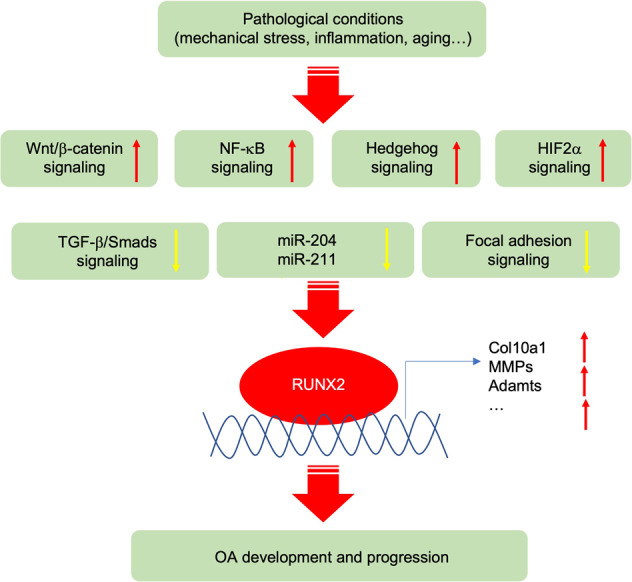
Osteoarthritis (OA) is a chronic degenerative joint disorder that leads to disability and affects more than 500 million population worldwide. OA was believed to be caused by the wearing and tearing of articular cartilage, but it is now more commonly referred to as a chronic whole-joint disorder that is initiated with biochemical and cellular alterations in the synovial joint tissues, which leads to the histological and structural changes of the joint and ends up with the whole tissue dysfunction. Currently, there is no cure for OA, partly due to a lack of comprehensive understanding of the pathological mechanism of the initiation and progression of the disease. Therefore, a better understanding of pathological signaling pathways and key molecules involved in OA pathogenesis is crucial for therapeutic target design and drug development. In this review, we first summarize the epidemiology of OA, including its prevalence, incidence and burdens, and OA risk factors. We then focus on the roles and regulation of the pathological signaling pathways, such as Wnt/β-catenin, NF-κB, focal adhesion, HIFs, TGFβ/ΒΜP and FGF signaling pathways, and key regulators AMPK, mTOR, and RUNX2 in the onset and development of OA. In addition, the roles of factors associated with OA, including MMPs, ADAMTS/ADAMs, and PRG4, are discussed in detail. Finally, we provide updates on the current clinical therapies and clinical trials of biological treatments and drugs for OA. Research advances in basic knowledge of articular cartilage biology and OA pathogenesis will have a significant impact and translational value in developing OA therapeutic strategies.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
