

Sulfamate Acetamides as Self-Immolative Electrophiles for Covalent Ligand-Directed Release Chemistry
- PMID: 36738297
- PMCID: PMC9936582
- DOI: 10.1021/jacs.2c08853
Sulfamate Acetamides as Self-Immolative Electrophiles for Covalent Ligand-Directed Release Chemistry
Abstract
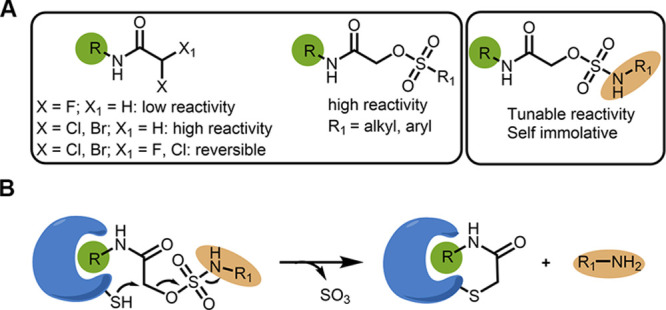
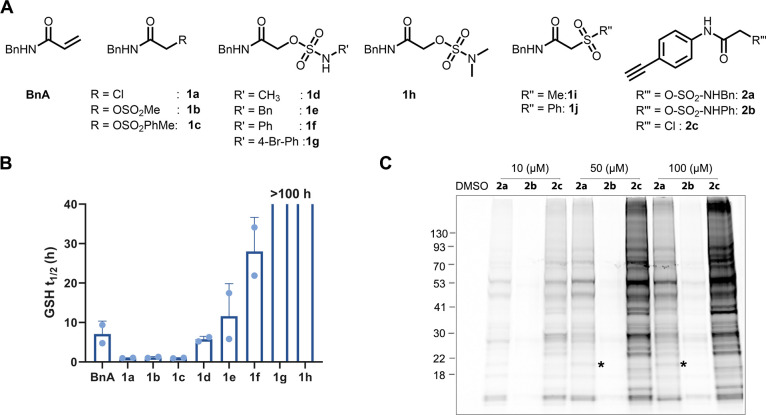
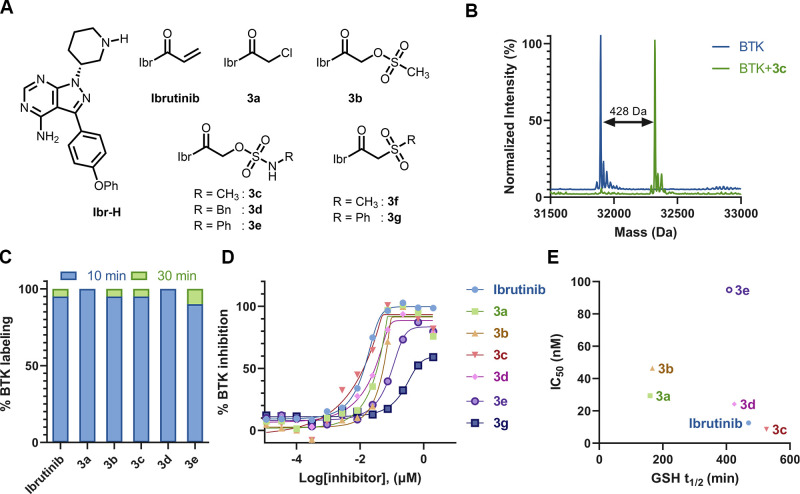
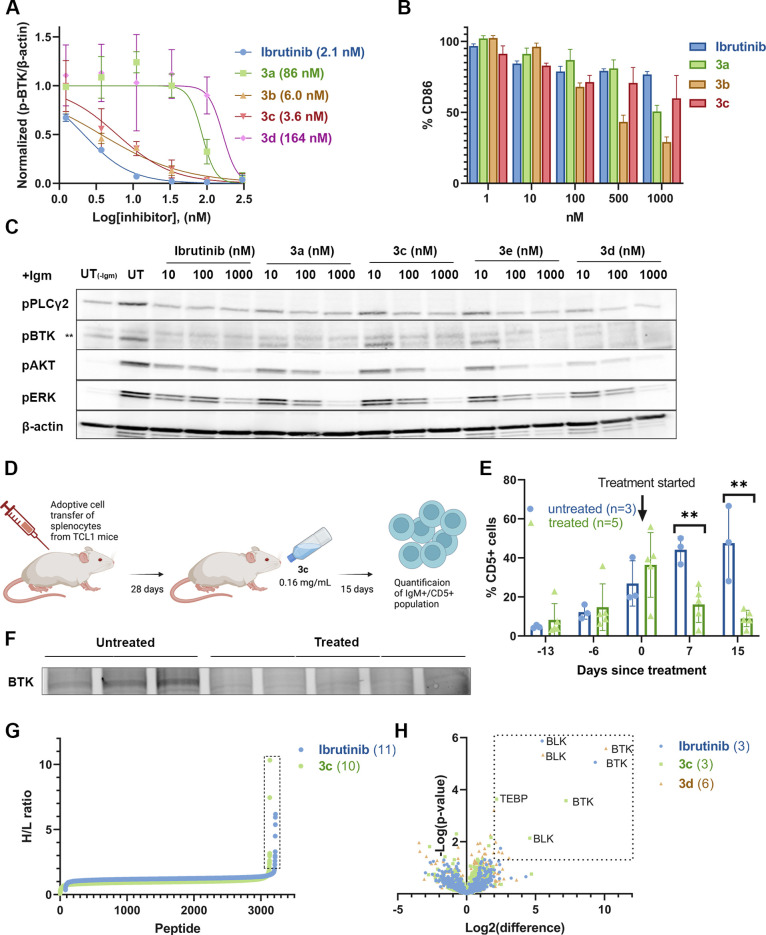
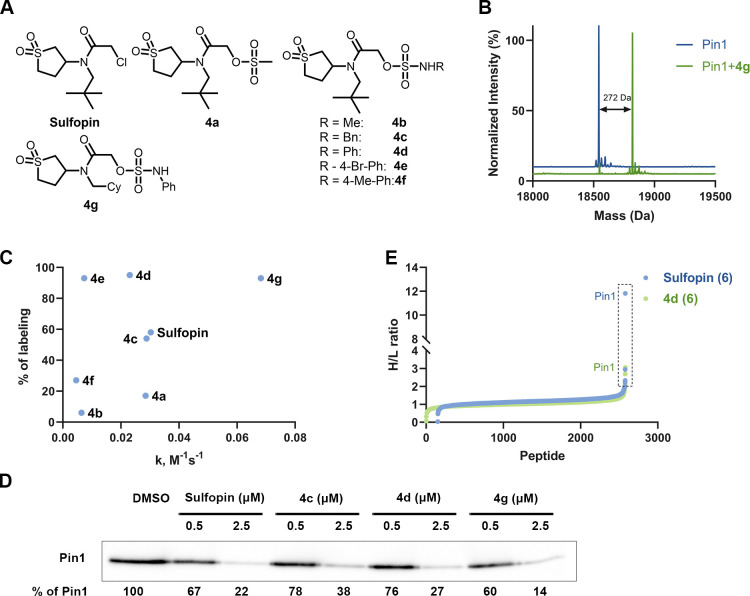
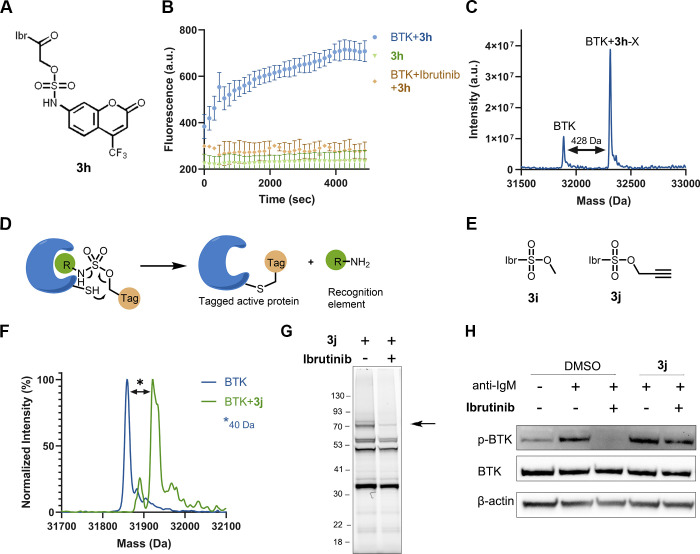
Electrophiles for covalent inhibitors that are suitable for in vivo administration are rare. While acrylamides are prevalent in FDA-approved covalent drugs, chloroacetamides are considered too reactive for such purposes. We report sulfamate-based electrophiles that maintain chloroacetamide-like geometry with tunable reactivity. In the context of the BTK inhibitor ibrutinib, sulfamate analogues showed low reactivity with comparable potency in protein labeling, in vitro, and cellular kinase activity assays and were effective in a mouse model of CLL. In a second example, we converted a chloroacetamide Pin1 inhibitor to a potent and selective sulfamate acetamide with improved buffer stability. Finally, we show that sulfamate acetamides can be used for covalent ligand-directed release (CoLDR) chemistry, both for the generation of "turn-on" probes as well as for traceless ligand-directed site-specific labeling of proteins. Taken together, this chemistry represents a promising addition to the list of electrophiles suitable for in vivo covalent targeting.
Conflict of interest statement
The authors declare the following competing financial interest(s): N.L. and R.N.R. are inventors on a provisional patent describing this technology.
Figures
References
-
- Backus K. M.; Correia B. E.; Lum K. M.; Forli S.; Horning B. D.; González-Páez G. E.; Chatterjee S.; Lanning B. R.; Teijaro J. R.; Olson A. J.; Wolan D. W.; Cravatt B. F. Proteome-Wide Covalent Ligand Discovery in Native Biological Systems. Nature 2016, 534, 570–574. 10.1038/nature18002. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
