

Aberrant JAK-STAT signaling-mediated chromatin remodeling impairs the sensitivity of NK/T-cell lymphoma to chidamide
- PMID: 36740715
- PMCID: PMC9900953
- DOI: 10.1186/s13148-023-01436-6
Aberrant JAK-STAT signaling-mediated chromatin remodeling impairs the sensitivity of NK/T-cell lymphoma to chidamide
Abstract
Background: Natural killer/T-cell lymphoma (NKTL) is a rare type of aggressive and heterogeneous non-Hodgkin's lymphoma (NHL) with a poor prognosis and limited therapeutic options. Therefore, there is an urgent need to exploit potential novel therapeutic targets for the treatment of NKTL. Histone deacetylase (HDAC) inhibitor chidamide was recently approved for treating relapsed/refractory peripheral T-cell lymphoma (PTCL) patients. However, its therapeutic efficacy in NKTL remains unclear.
Methods: We performed a phase II clinical trial to evaluate the efficacy of chidamide in 28 relapsed/refractory NKTL patients. Integrative transcriptomic, chromatin profiling analysis and functional studies were performed to identify potential predictive biomarkers and unravel the mechanisms of resistance to chidamide. Immunohistochemistry (IHC) was used to validate the predictive biomarkers in tumors from the clinical trial.
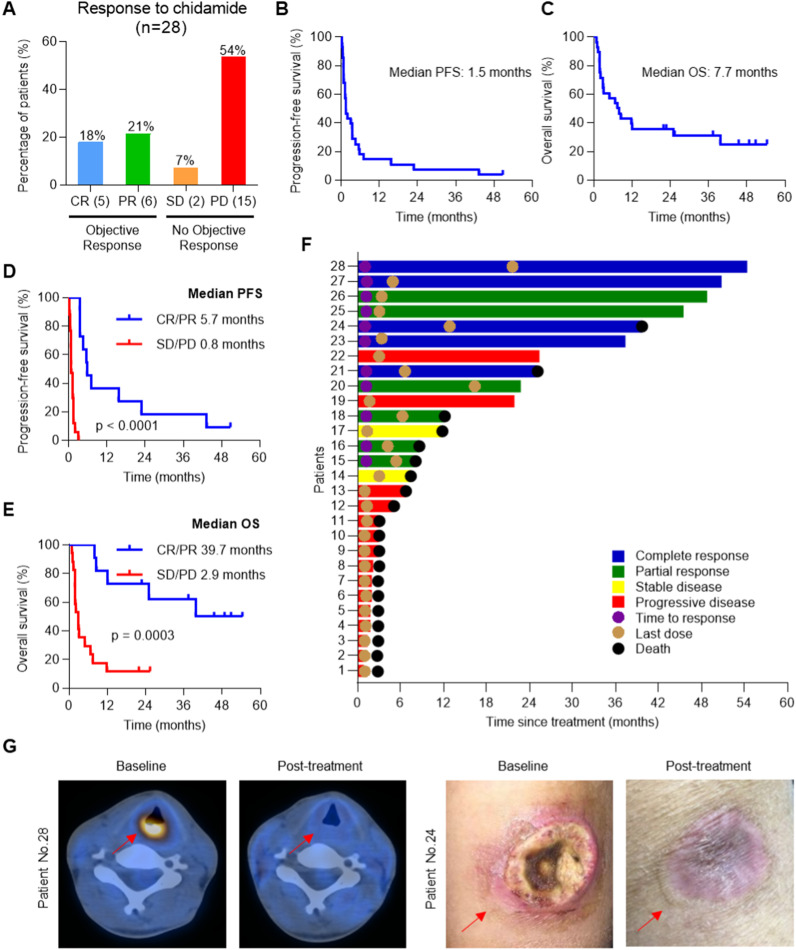
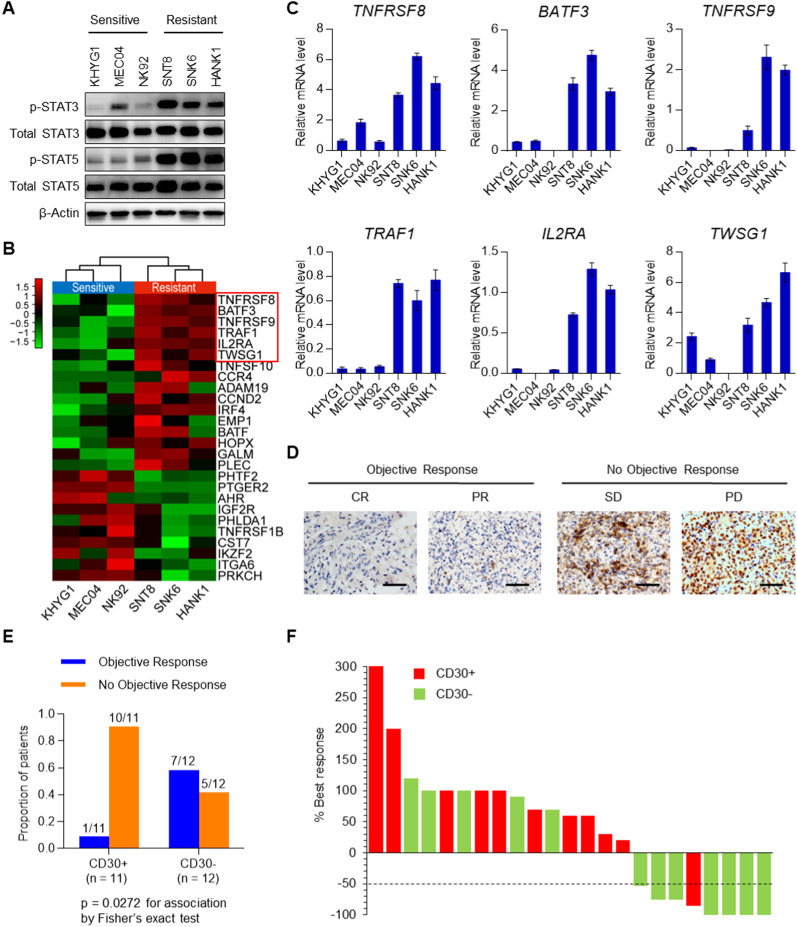
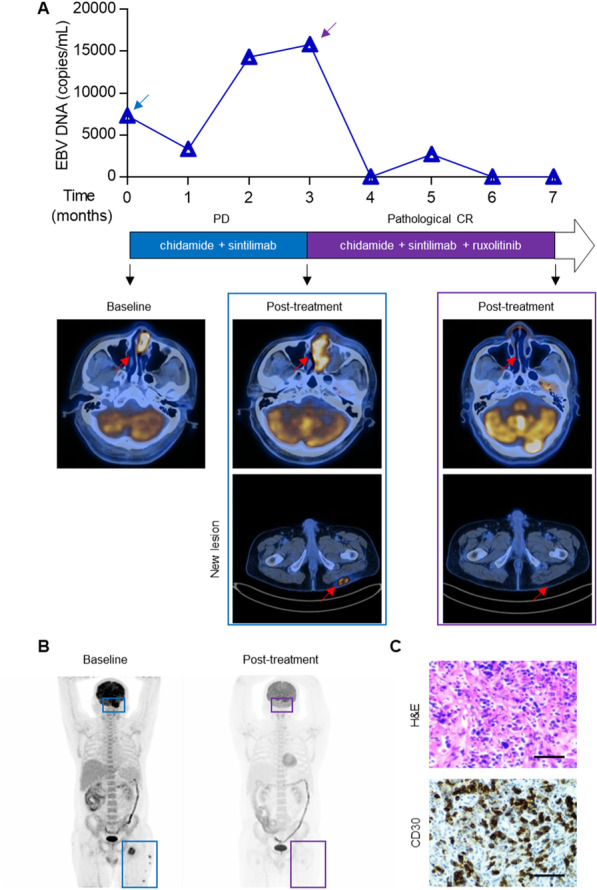
Results: We demonstrated that chidamide is effective in treating relapsed/refractory NKTL patients, achieving an overall response and complete response rate of 39 and 18%, respectively. In vitro studies showed that hyperactivity of JAK-STAT signaling in NKTL cell lines was associated with the resistance to chidamide. Mechanistically, our results revealed that aberrant JAK-STAT signaling remodels the chromatin and confers resistance to chidamide. Subsequently, inhibition of JAK-STAT activity could overcome resistance to chidamide by reprogramming the chromatin from a resistant to sensitive state, leading to synergistic anti-tumor effect in vitro and in vivo. More importantly, our clinical data demonstrated that combinatorial therapy with chidamide and JAK inhibitor ruxolitinib is effective against chidamide-resistant NKTL. In addition, we identified TNFRSF8 (CD30), a downstream target of the JAK-STAT pathway, as a potential biomarker that could predict NKTL sensitivity to chidamide.
Conclusions: Our study suggests that chidamide, in combination with JAK-STAT inhibitors, can be a novel targeted therapy in the standard of care for NKTL.
Trial registration: ClinicalTrials.gov, NCT02878278. Registered 25 August 2016, https://clinicaltrials.gov/ct2/show/NCT02878278.
Keywords: Chidamide resistance; Chromatin remodeling; HDAC inhibitor; JAK-STAT pathway; NKTL.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. - PubMed
-
- Gualco G, Domeny-Duarte P, Chioato L, Barber G, Natkunam Y, Bacchi CE. Clinicopathologic and molecular features of 122 Brazilian cases of nodal and extranodal NK/T-cell lymphoma, nasal type, with EBV subtyping analysis. Am J Surg Pathol. 2011;35(8):1195–1203. - PubMed
-
- Suzuki R, Takeuchi K, Ohshima K, Nakamura S. Extranodal NK/T-cell lymphoma: diagnosis and treatment cues. Hematol Oncol. 2008;26(2):66–72. - PubMed
-
- Liang R. Advances in the management and monitoring of extranodal NK/T-cell lymphoma, nasal type. Br J Haematol. 2009;147(1):13–21. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
