

Airway microbiome-immune crosstalk in chronic obstructive pulmonary disease
- PMID: 36741369
- PMCID: PMC9890194
- DOI: 10.3389/fimmu.2022.1085551
Airway microbiome-immune crosstalk in chronic obstructive pulmonary disease
Abstract
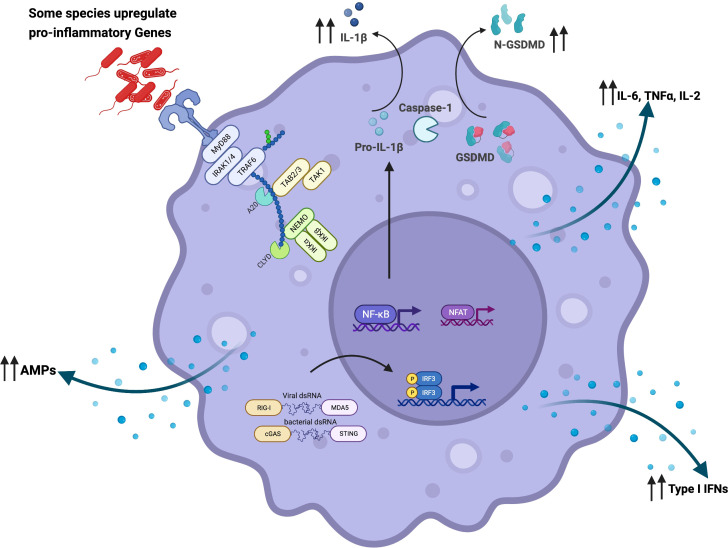
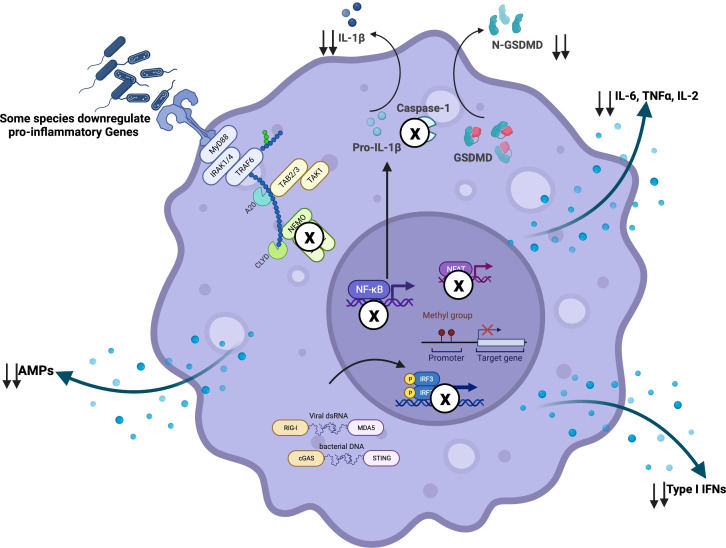
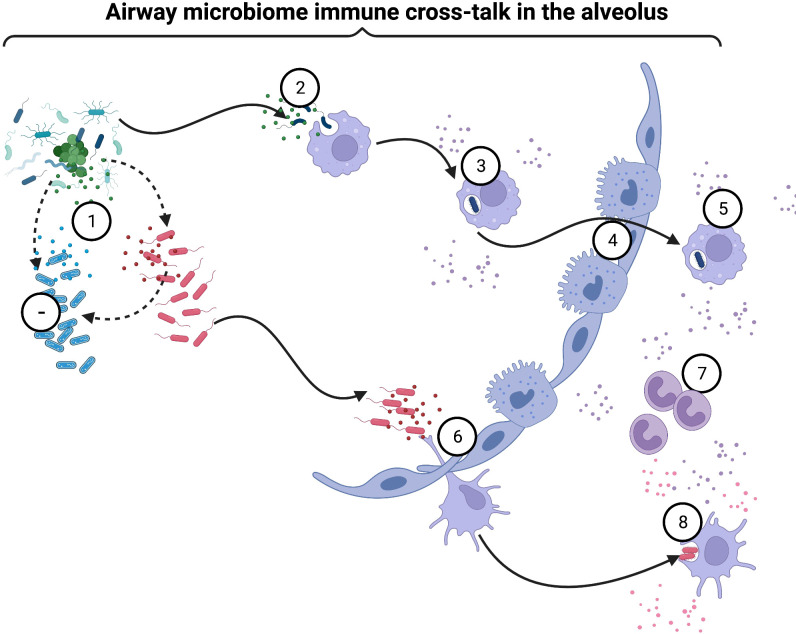
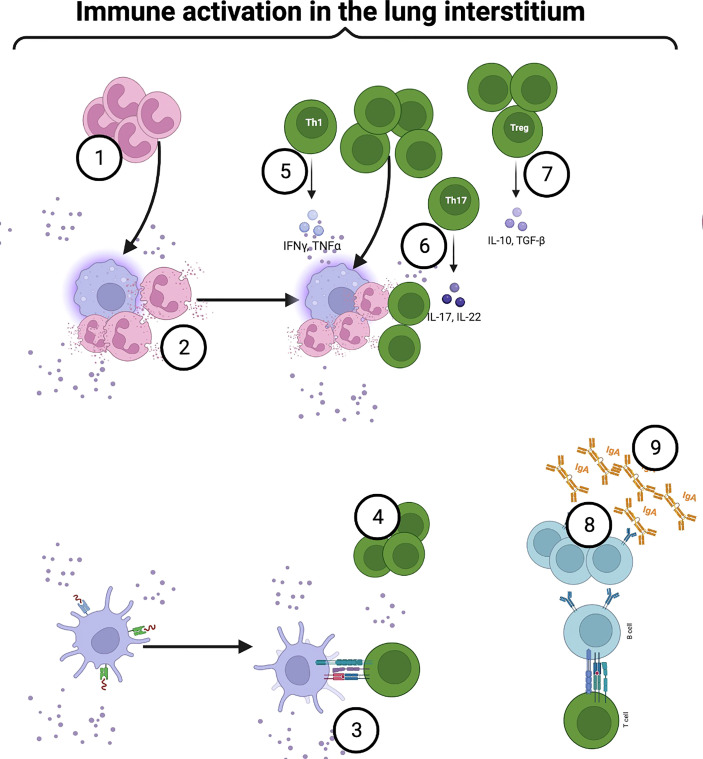
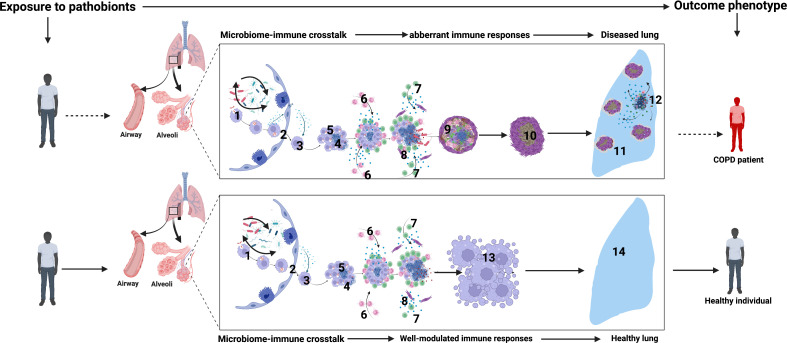
Chronic Obstructive Pulmonary Disease (COPD) has significantly contributed to global mortality, with three million deaths reported annually. This impact is expected to increase over the next 40 years, with approximately 5 million people predicted to succumb to COPD-related deaths annually. Immune mechanisms driving disease progression have not been fully elucidated. Airway microbiota have been implicated. However, it is still unclear how changes in the airway microbiome drive persistent immune activation and consequent lung damage. Mechanisms mediating microbiome-immune crosstalk in the airways remain unclear. In this review, we examine how dysbiosis mediates airway inflammation in COPD. We give a detailed account of how airway commensal bacteria interact with the mucosal innate and adaptive immune system to regulate immune responses in healthy or diseased airways. Immune-phenotyping airway microbiota could advance COPD immunotherapeutics and identify key open questions that future research must address to further such translation.
Keywords: COPD; adaptive immunity; inflammation; innate immunity; lung microbiome; mucosal immunity.
Copyright © 2023 Kayongo, Robertson, Siddharthan, Ntayi, Ndawula, Sande, Bagaya, Kirenga, Mayanja-Kizza, Joloba and Forslund.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Barbu C, Iordache M, Man M. Inflammation in COPD: pathogenesis, local and systemic effects. Rom J Morphol Embryol (2011) 52(1):21–7. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical