

Review
doi: 10.1039/d2sc05089g.
eCollection 2023 Jan 4.
Predictive chemistry: machine learning for reaction deployment, reaction development, and reaction discovery
Affiliations
- PMID: 36743887
- PMCID: PMC9811563
- DOI: 10.1039/d2sc05089g
Item in Clipboard
Review
Predictive chemistry: machine learning for reaction deployment, reaction development, and reaction discovery
Chem Sci.
.
Abstract
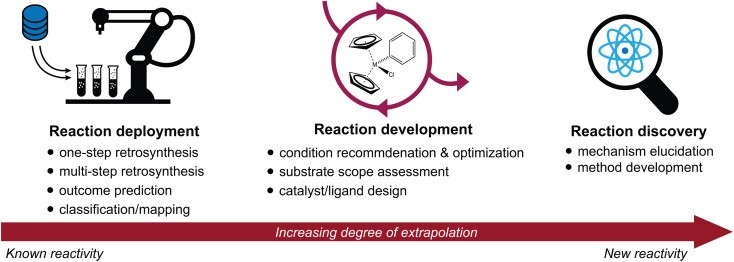
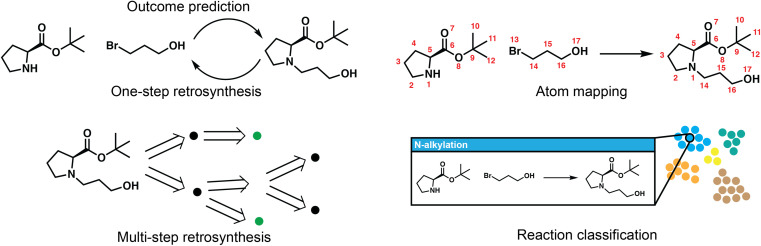
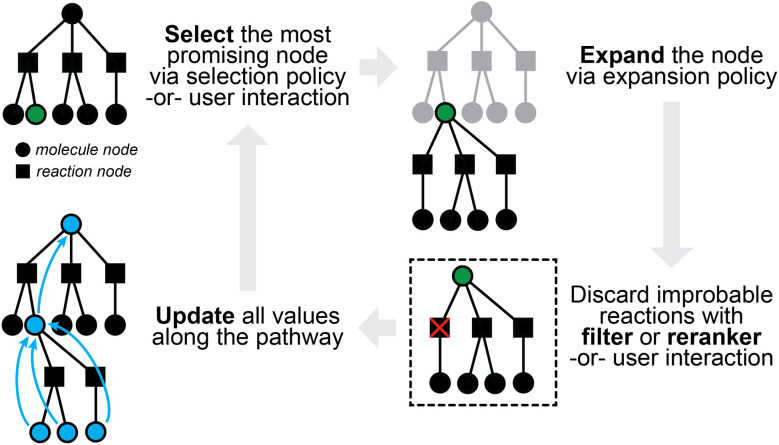
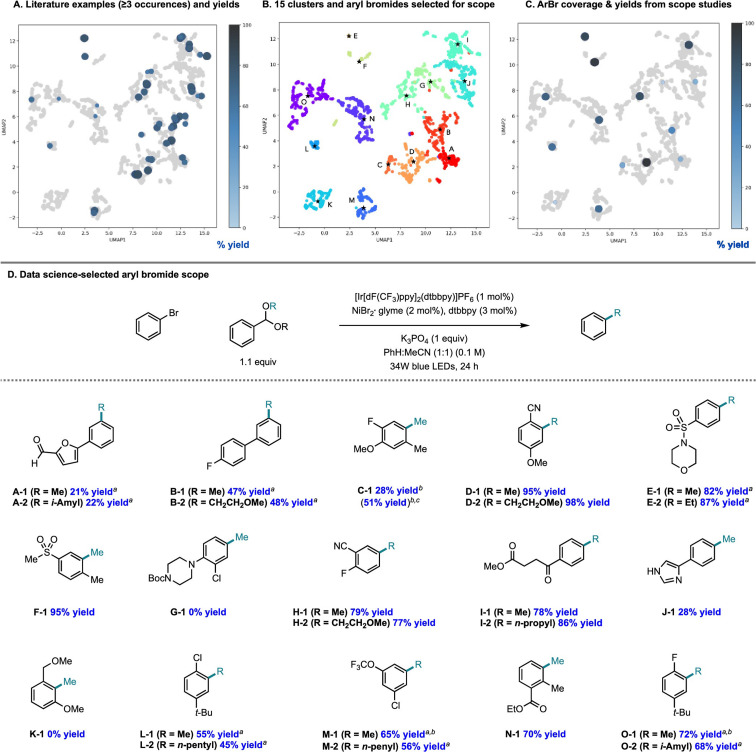
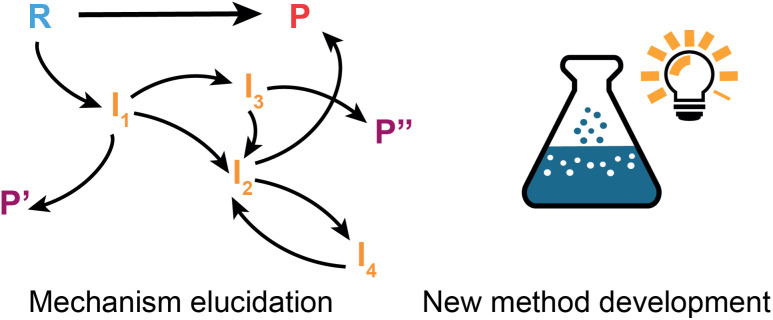
The field of predictive chemistry relates to the development of models able to describe how molecules interact and react. It encompasses the long-standing task of computer-aided retrosynthesis, but is far more reaching and ambitious in its goals. In this review, we summarize several areas where predictive chemistry models hold the potential to accelerate the deployment, development, and discovery of organic reactions and advance synthetic chemistry.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
Authors declare no competing interests.
Figures




Similar articles
-
Chemistry-informed molecular graph as reaction descriptor for machine-learned retrosynthesis planning.Proc Natl Acad Sci U S A. 2022 Oct 11;119(41):e2212711119. doi: 10.1073/pnas.2212711119. Epub 2022 Oct 3. Proc Natl Acad Sci U S A. 2022. PMID: 36191228 Free PMC article.
-
RetroComposer: Composing Templates for Template-Based Retrosynthesis Prediction.Biomolecules. 2022 Sep 19;12(9):1325. doi: 10.3390/biom12091325. Biomolecules. 2022. PMID: 36139164 Free PMC article.
-
Data-driven Chemical Reaction Prediction and Retrosynthesis.Chimia (Aarau). 2019 Dec 18;73(12):997-1000. doi: 10.2533/chimia.2019.997. Chimia (Aarau). 2019. PMID: 31883550
-
De Novo Peptide and Protein Design Using Generative Adversarial Networks: An Update.J Chem Inf Model. 2022 Feb 28;62(4):761-774. doi: 10.1021/acs.jcim.1c01361. Epub 2022 Feb 7. J Chem Inf Model. 2022. PMID: 35128926 Review.
-
Deep learning in retrosynthesis planning: datasets, models and tools.Brief Bioinform. 2022 Jan 17;23(1):bbab391. doi: 10.1093/bib/bbab391. Brief Bioinform. 2022. PMID: 34571535 Review.
Cited by
-
Dataset Design for Building Models of Chemical Reactivity.ACS Cent Sci. 2023 Dec 8;9(12):2196-2204. doi: 10.1021/acscentsci.3c01163. eCollection 2023 Dec 27. ACS Cent Sci. 2023. PMID: 38161380 Free PMC article. Review.
-
Enabling high throughput deep reinforcement learning with first principles to investigate catalytic reaction mechanisms.Nat Commun. 2024 Jul 25;15(1):6281. doi: 10.1038/s41467-024-50531-6. Nat Commun. 2024. PMID: 39060277 Free PMC article.
-
Fine-tuning GPT-3 for machine learning electronic and functional properties of organic molecules.Chem Sci. 2023 Dec 5;15(2):500-510. doi: 10.1039/d3sc04610a. eCollection 2024 Jan 3. Chem Sci. 2023. PMID: 38179524 Free PMC article.
-
Open-Source Machine Learning in Computational Chemistry.J Chem Inf Model. 2023 Aug 14;63(15):4505-4532. doi: 10.1021/acs.jcim.3c00643. Epub 2023 Jul 19. J Chem Inf Model. 2023. PMID: 37466636 Free PMC article. Review.
-
LinChemIn: SynGraph-a data model and a toolkit to analyze and compare synthetic routes.J Cheminform. 2023 Apr 1;15(1):41. doi: 10.1186/s13321-023-00714-y. J Cheminform. 2023. PMID: 37005691 Free PMC article.
References
-
- de Almeida A. F. Moreira R. Rodrigues T. Nat. Rev. Chem. 2019;3:589–604. doi: 10.1038/s41570-019-0124-0. - DOI
-
- Schwaller P. Vaucher A. C. Laplaza R. Bunne C. Krause A. Corminboeuf C. Laino T. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2022:e1604.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources