

Dysregulated Smooth Muscle Cell BMPR2-ARRB2 Axis Causes Pulmonary Hypertension
- PMID: 36744494
- PMCID: PMC10008520
- DOI: 10.1161/CIRCRESAHA.121.320541
Dysregulated Smooth Muscle Cell BMPR2-ARRB2 Axis Causes Pulmonary Hypertension
Abstract
Objective: Mutations in BMPR2 (bone morphogenetic protein receptor 2) are associated with familial and sporadic pulmonary arterial hypertension (PAH). The functional and molecular link between loss of BMPR2 in pulmonary artery smooth muscle cells (PASMC) and PAH pathogenesis warrants further investigation, as most investigations focus on BMPR2 in pulmonary artery endothelial cells. Our goal was to determine whether and how decreased BMPR2 is related to the abnormal phenotype of PASMC in PAH.
Methods: SMC-specific Bmpr2-/- mice (BKOSMC) were created and compared to controls in room air, after 3 weeks of hypoxia as a second hit, and following 4 weeks of normoxic recovery. Echocardiography, right ventricular systolic pressure, and right ventricular hypertrophy were assessed as indices of pulmonary hypertension. Proliferation, contractility, gene and protein expression of PASMC from BKOSMC mice, human PASMC with BMPR2 reduced by small interference RNA, and PASMC from PAH patients with a BMPR2 mutation were compared to controls, to investigate the phenotype and underlying mechanism.
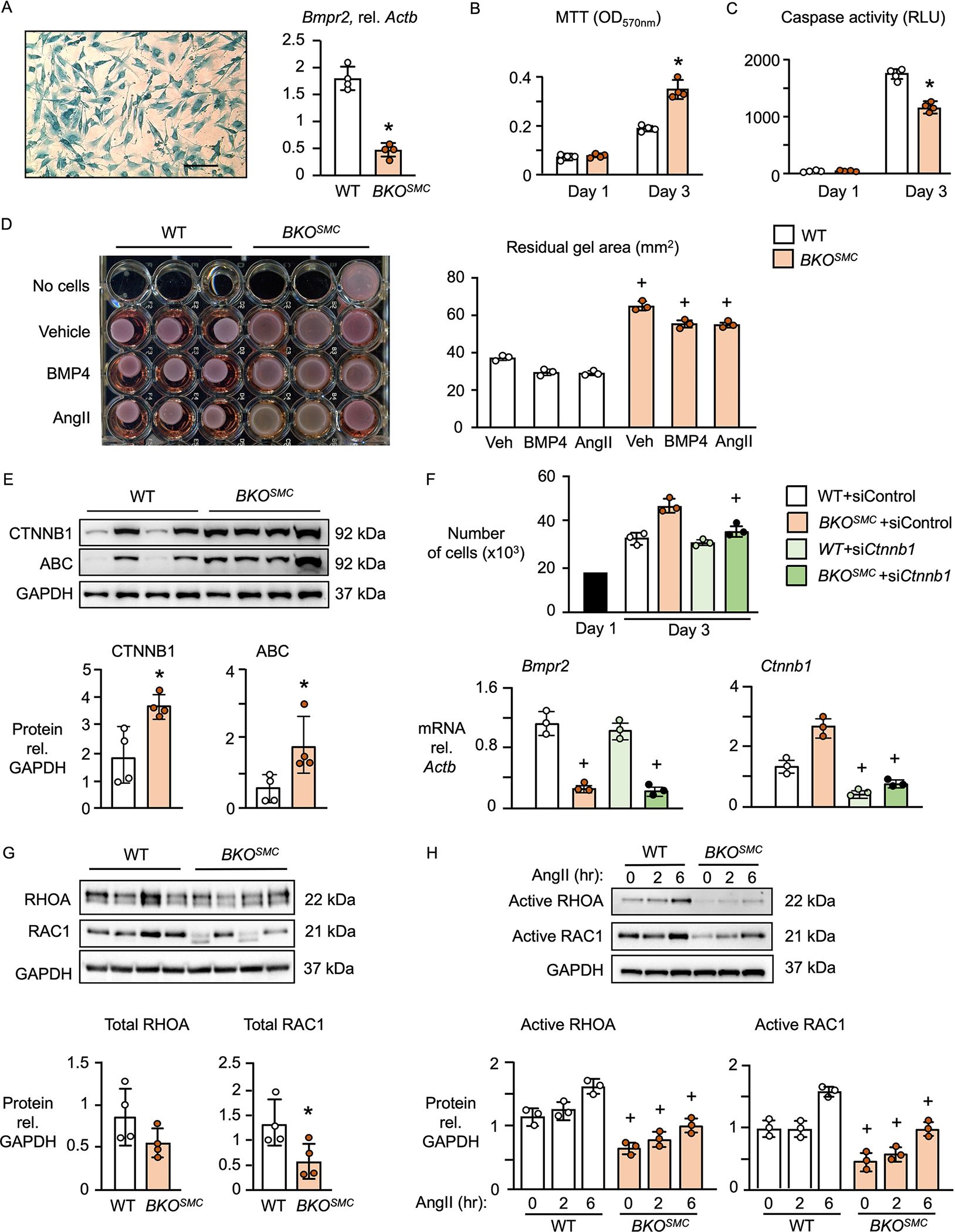
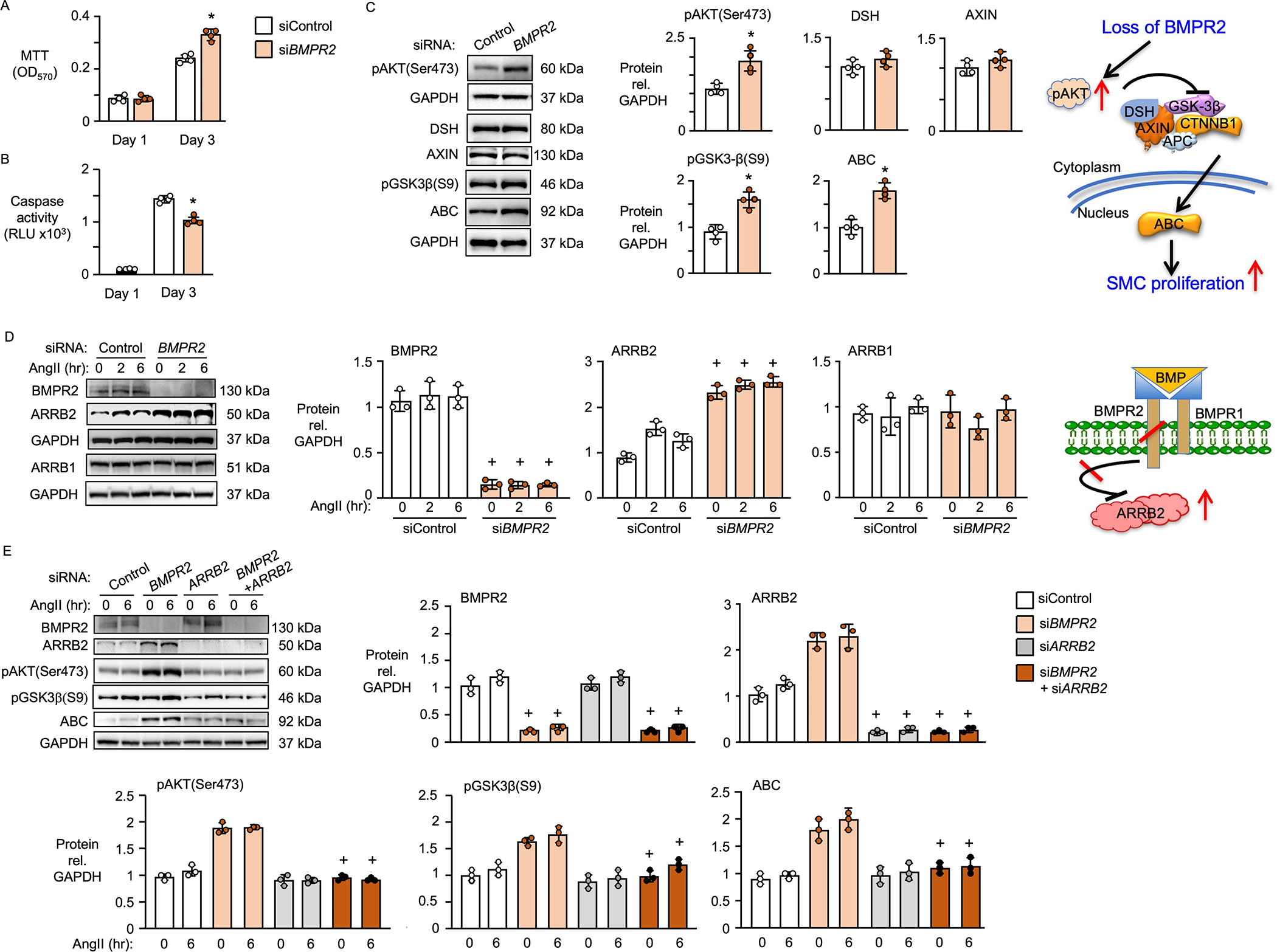
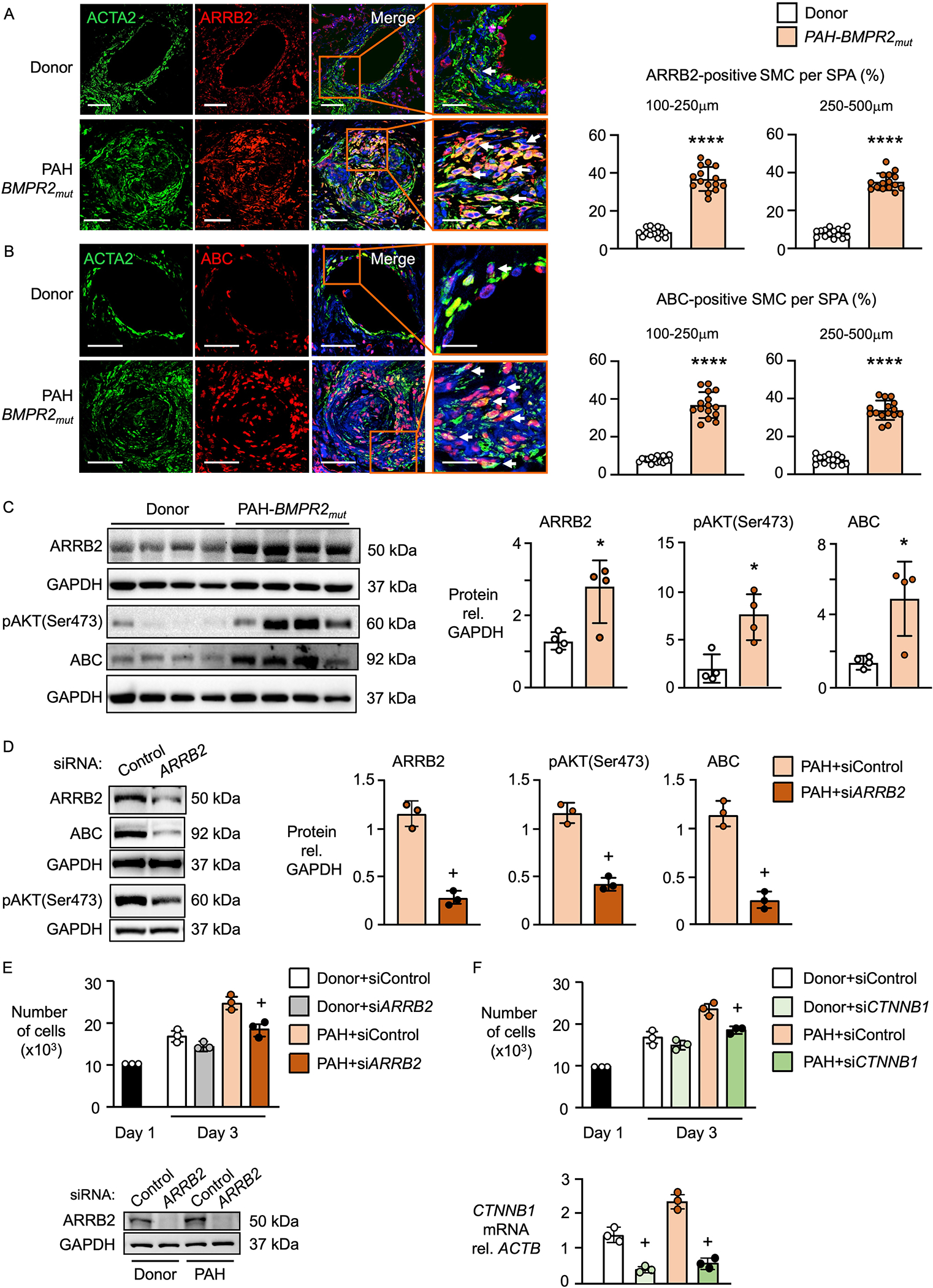
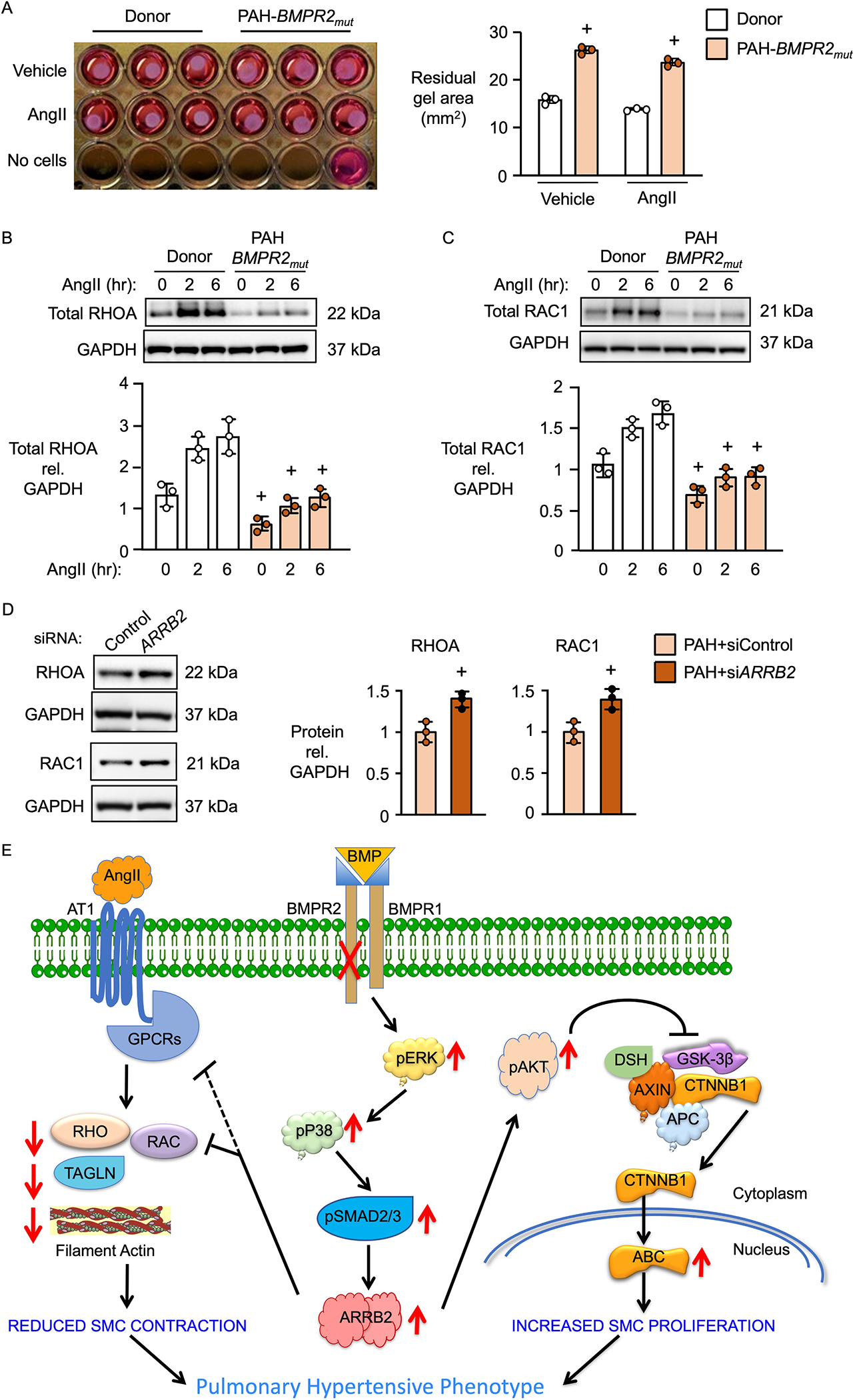
Results: BKOSMC mice showed reduced hypoxia-induced vasoconstriction and persistent pulmonary hypertension following recovery from hypoxia, associated with sustained muscularization of distal pulmonary arteries. PASMC from mutant compared to control mice displayed reduced contractility at baseline and in response to angiotensin II, increased proliferation and apoptosis resistance. Human PASMC with reduced BMPR2 by small interference RNA, and PASMC from PAH patients with a BMPR2 mutation showed a similar phenotype related to upregulation of pERK1/2 (phosphorylated extracellular signal related kinase 1/2)-pP38-pSMAD2/3 mediating elevation in ARRB2 (β-arrestin2), pAKT (phosphorylated protein kinase B) inactivation of GSK3-beta, CTNNB1 (β-catenin) nuclear translocation and reduction in RHOA (Ras homolog family member A) and RAC1 (Ras-related C3 botulinum toxin substrate 1). Decreasing ARRB2 in PASMC with reduced BMPR2 restored normal signaling, reversed impaired contractility and attenuated heightened proliferation and in mice with inducible loss of BMPR2 in SMC, decreasing ARRB2 prevented persistent pulmonary hypertension.
Conclusions: Agents that neutralize the elevated ARRB2 resulting from loss of BMPR2 in PASMC could prevent or reverse the aberrant hypocontractile and hyperproliferative phenotype of these cells in PAH.
Keywords: beta-arrestin 2; bone morphogenetic protein receptors, type II; pulmonary arterial hypertension; smooth muscle cells; transgenic mice.
Figures





References
-
- Deng Z, Haghighi F, Helleby L, Vanterpool K, Horn EM, Barst RJ, Hodge SE, Morse JH, Knowles JA. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am J Respir Crit Care Med. 2000;161:1055–1059. - PubMed
-
- Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
