

Discovery of Targets for Immune-Metabolic Antitumor Drugs Identifies Estrogen-Related Receptor Alpha
- PMID: 36745048
- PMCID: PMC9975674
- DOI: 10.1158/2159-8290.CD-22-0244
Discovery of Targets for Immune-Metabolic Antitumor Drugs Identifies Estrogen-Related Receptor Alpha
Abstract
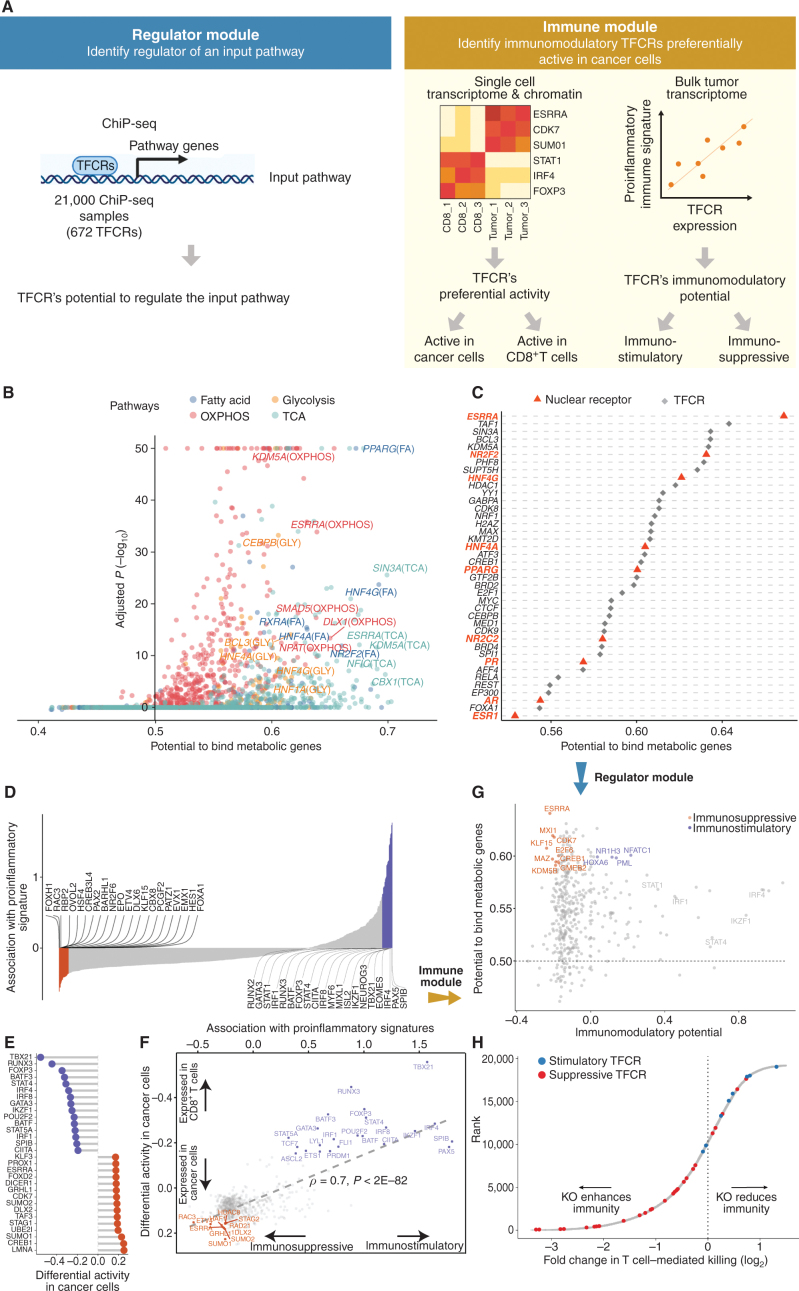
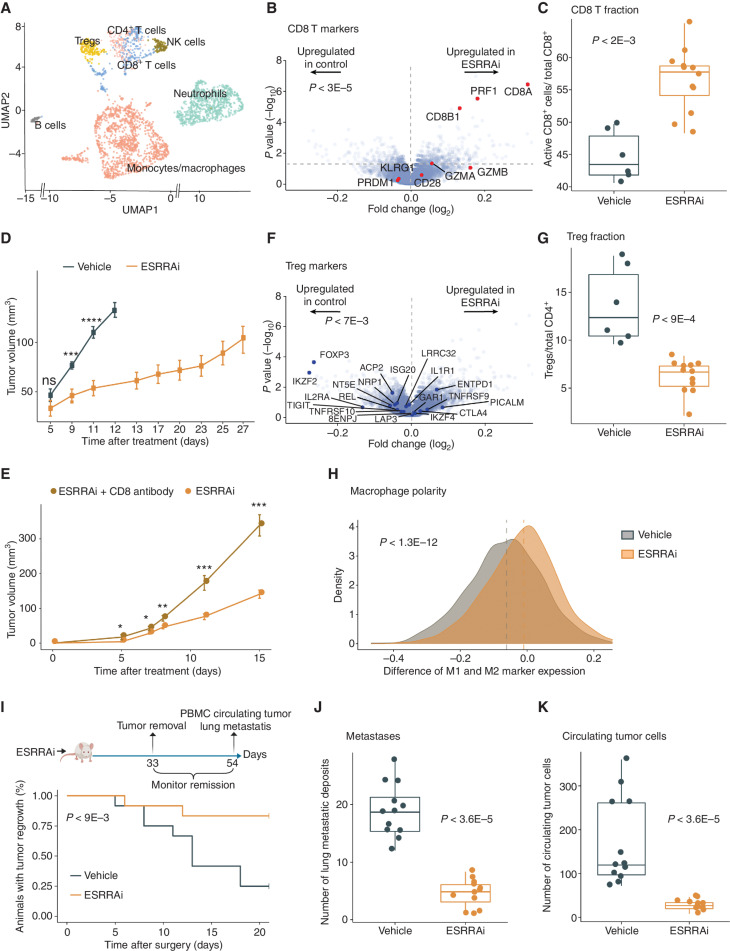
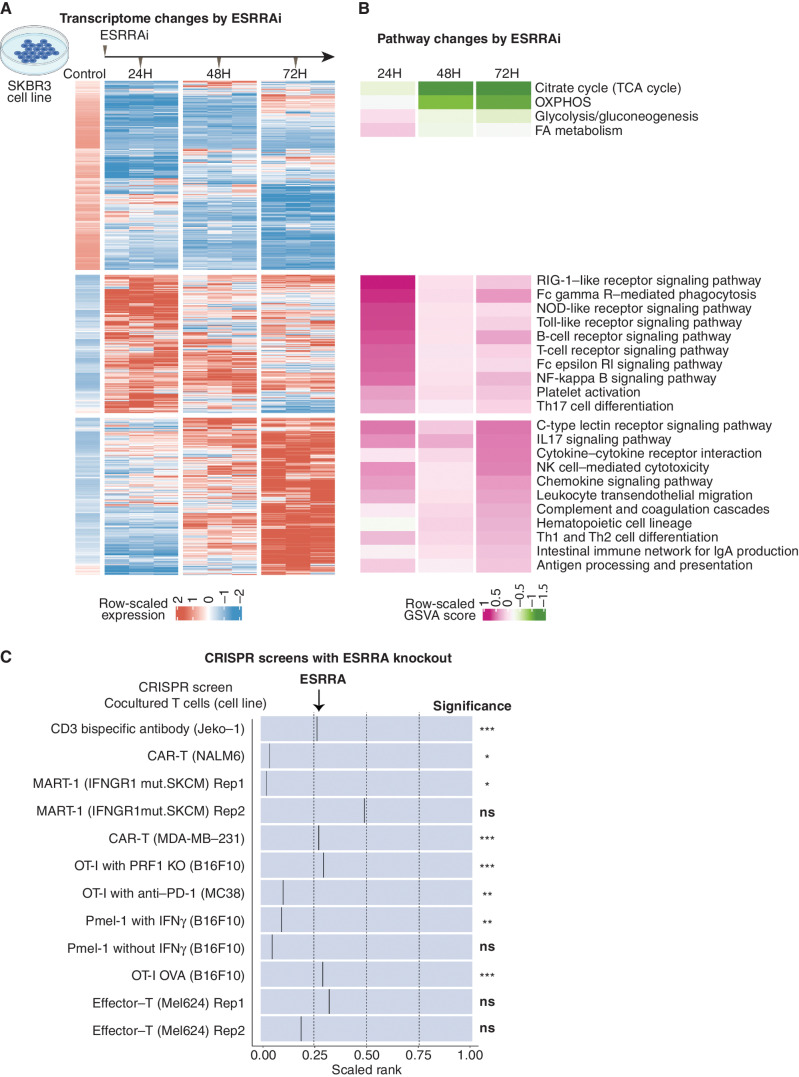
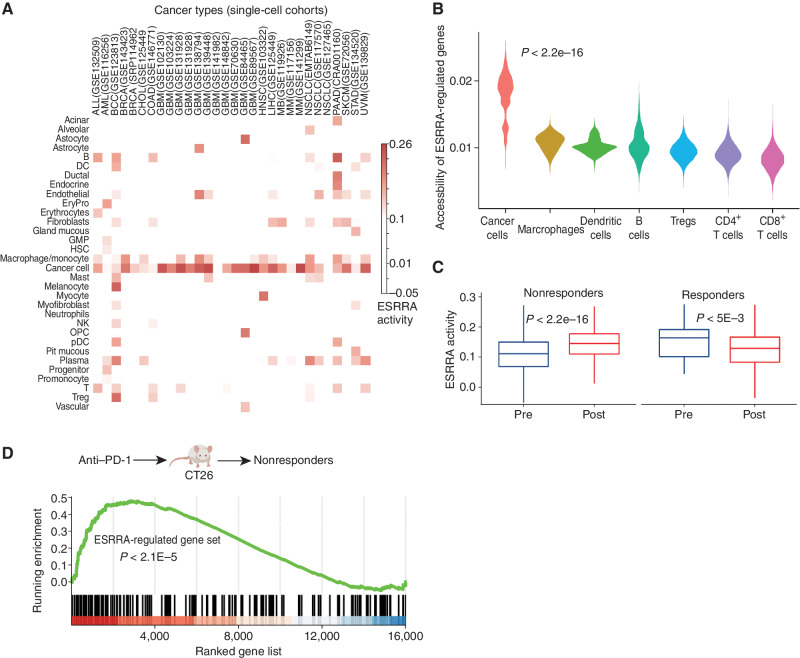
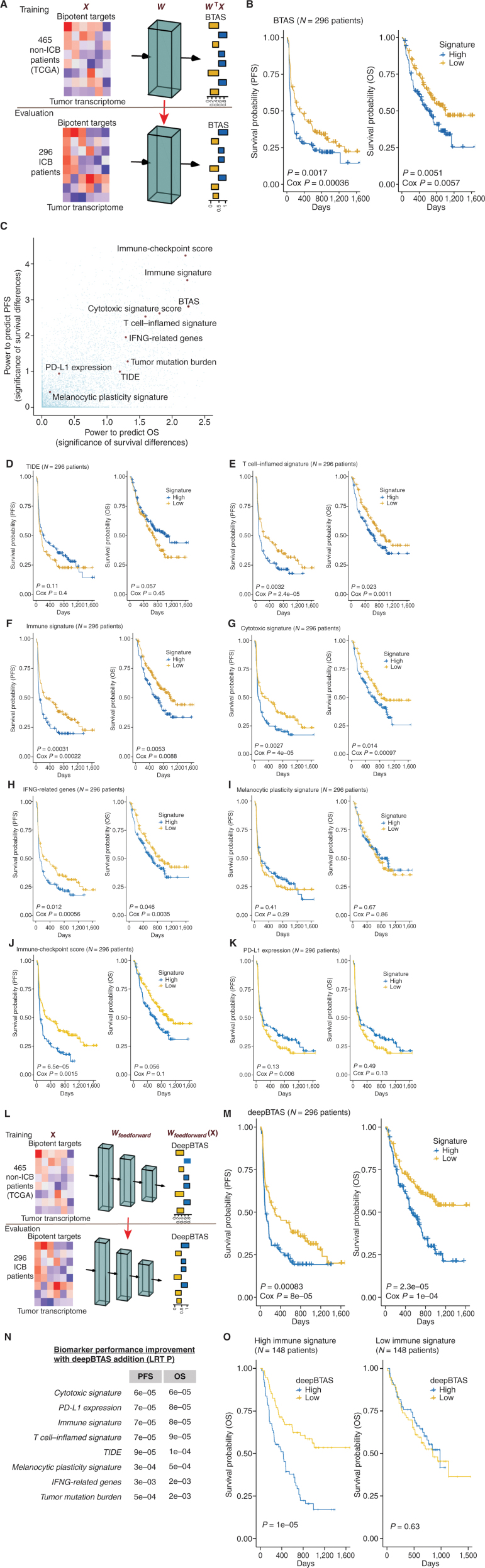
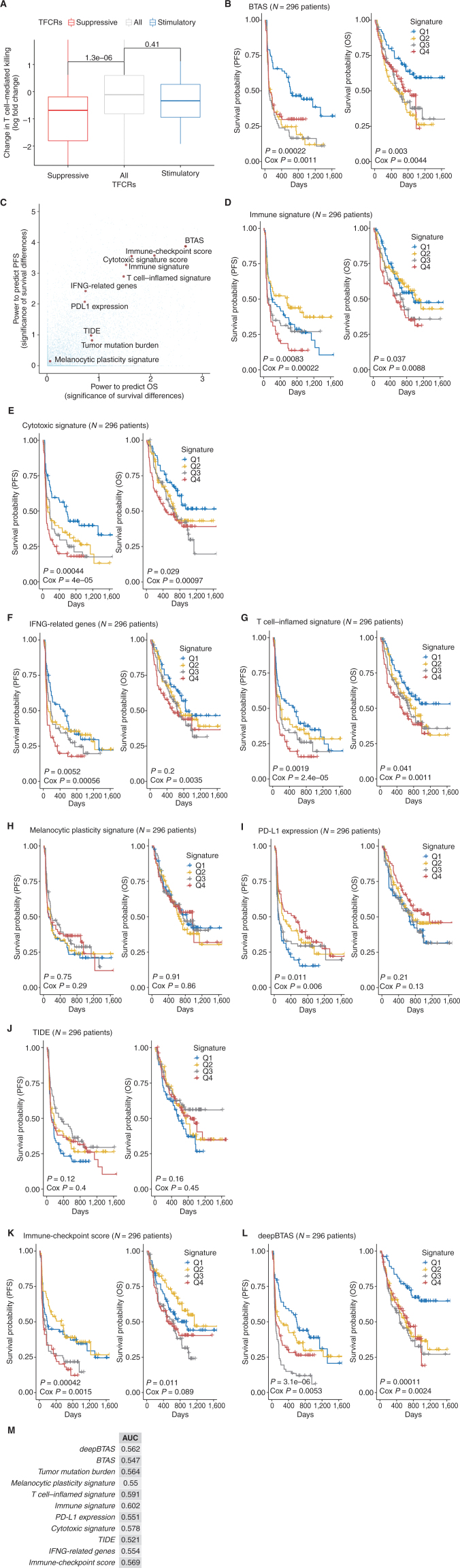
Drugs that kill tumors through multiple mechanisms have the potential for broad clinical benefits. Here, we first developed an in silico multiomics approach (BipotentR) to find cancer cell-specific regulators that simultaneously modulate tumor immunity and another oncogenic pathway and then used it to identify 38 candidate immune-metabolic regulators. We show the tumor activities of these regulators stratify patients with melanoma by their response to anti-PD-1 using machine learning and deep neural approaches, which improve the predictive power of current biomarkers. The topmost identified regulator, ESRRA, is activated in immunotherapy-resistant tumors. Its inhibition killed tumors by suppressing energy metabolism and activating two immune mechanisms: (i) cytokine induction, causing proinflammatory macrophage polarization, and (ii) antigen-presentation stimulation, recruiting CD8+ T cells into tumors. We also demonstrate a wide utility of BipotentR by applying it to angiogenesis and growth suppressor evasion pathways. BipotentR (http://bipotentr.dfci.harvard.edu/) provides a resource for evaluating patient response and discovering drug targets that act simultaneously through multiple mechanisms.
Significance: BipotentR presents resources for evaluating patient response and identifying targets for drugs that can kill tumors through multiple mechanisms concurrently. Inhibition of the topmost candidate target killed tumors by suppressing energy metabolism and effects on two immune mechanisms. This article is highlighted in the In This Issue feature, p. 517.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
- 2159-8274. doi: 10.1158/2159-8290.CD-13-3-ITI doi: 10.1158/2159-8290.CD-13-3-ITI
References
-
- Benesic A, Jalal K, Gerbes AL. Drug-drug combinations can enhance toxicity as shown by monocyte-derived hepatocyte-like cells from patients with idiosyncratic drug-induced liver injury. Toxicol Sci 2019;171:296–302. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
