

Innate sensing of picornavirus infection involves cGAS-STING-mediated antiviral responses triggered by mitochondrial DNA release
- PMID: 36745686
- PMCID: PMC9934381
- DOI: 10.1371/journal.ppat.1011132
Innate sensing of picornavirus infection involves cGAS-STING-mediated antiviral responses triggered by mitochondrial DNA release
Abstract
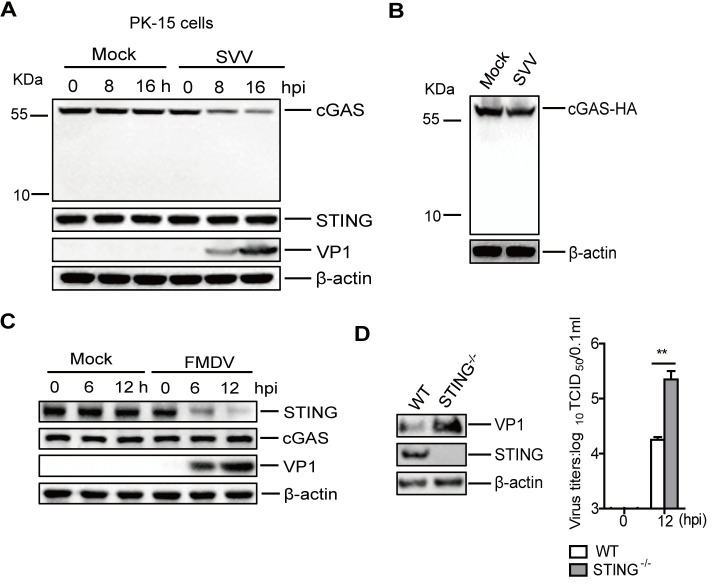
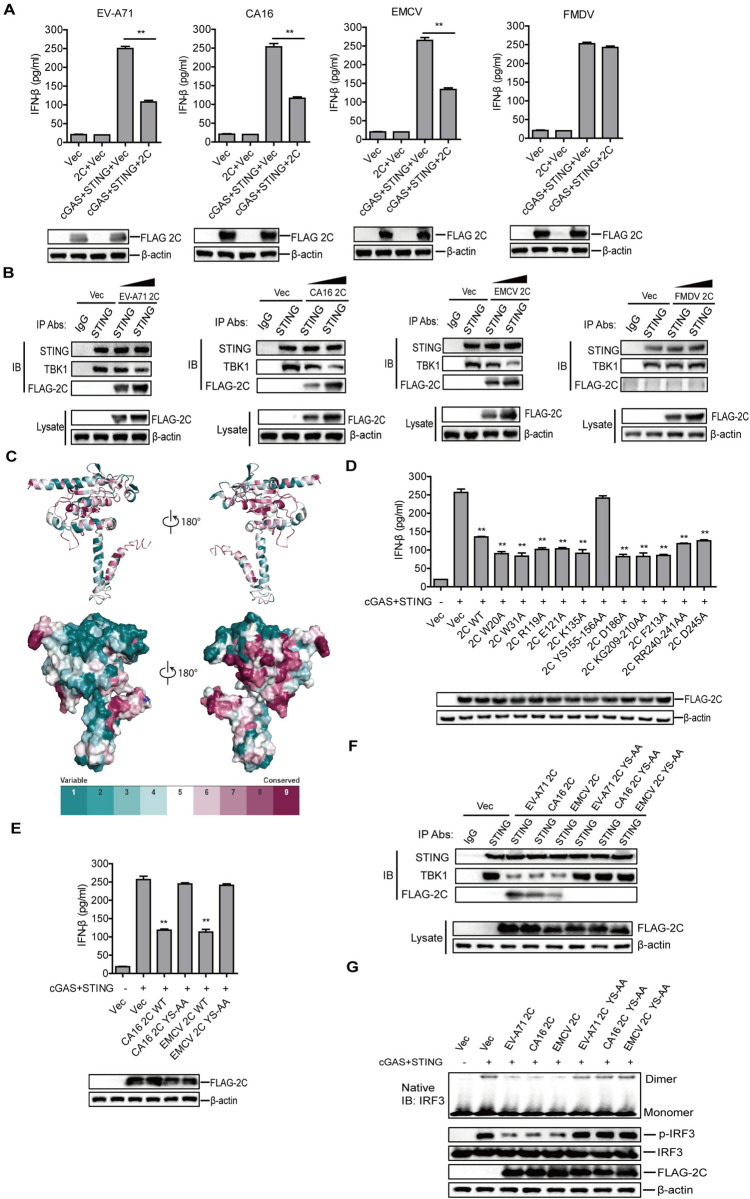
Cyclic GMP-AMP synthase (cGAS) plays a key role in the innate immune responses to both DNA and RNA virus infection. Here, we found that enterovirus 71 (EV-A71), Seneca Valley virus (SVV), and foot-and-mouth disease virus (FMDV) infection triggered mitochondria damage and mitochondrial DNA (mtDNA) release in vitro and vivo. These responses were mediated by picornavirus 2B proteins which induced mtDNA release during viral replication. SVV infection caused the opening of mitochondrial permeability transition pore (mPTP) and led to voltage-dependent anion channel 1 (VDAC1)- and BCL2 antagonist/killer 1 (Bak) and Bak/BCL2-associated X (Bax)-dependent mtDNA leakage into the cytoplasm, while EV-A71 and FMDV infection induced mPTP opening and resulted in VDAC1-dependent mtDNA release. The released mtDNA bound to cGAS and activated cGAS-mediated antiviral immune response. cGAS was essential for inhibiting EV-A71, SVV, and FMDV replication by regulation of IFN-β production. cGAS deficiency contributed to higher mortality of EV-A71- or FMDV-infected mice. In addition, we found that SVV 2C protein was responsible for decreasing cGAS expression through the autophagy pathway. The 9th and 153rd amino acid sites in 2C were critical for induction of cGAS degradation. Furthermore, we also show that EV-A71, CA16, and EMCV 2C antagonize the cGAS-stimulator of interferon genes (STING) pathway through interaction with STING, and highly conserved amino acids Y155 and S156 were critical for this inhibitory effect. In conclusion, these data reveal novel mechanisms of picornaviruses to block the antiviral effect mediated by the cGAS-STING signaling pathway, which will provide insights for developing antiviral strategies against picornaviruses.
Copyright: © 2023 Liu et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures









References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
