

This is a preprint.
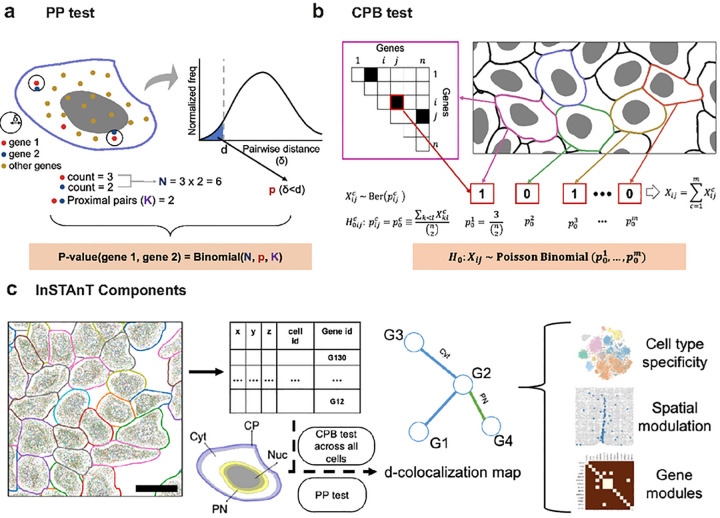
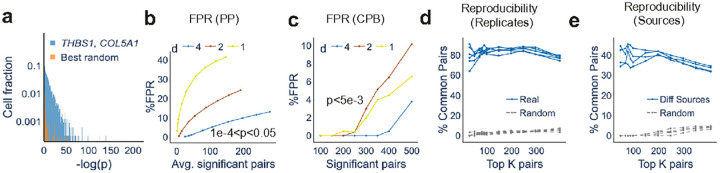
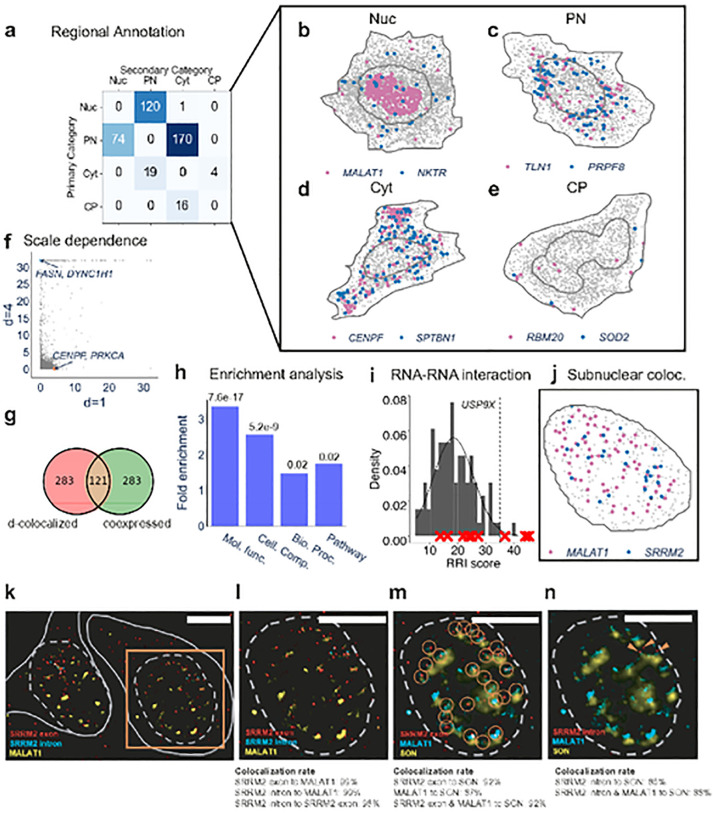
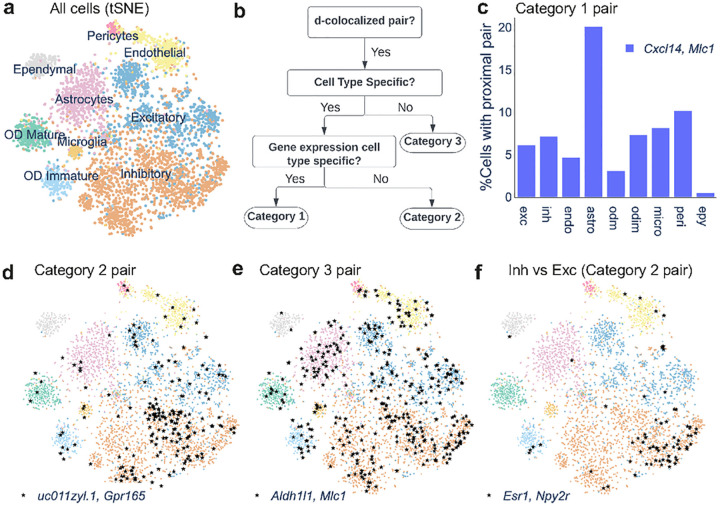
Intracellular Spatial Transcriptomic Analysis Toolkit (InSTAnT)
- PMID: 36747718
- PMCID: PMC9901031
- DOI: 10.21203/rs.3.rs-2481749/v1
Intracellular Spatial Transcriptomic Analysis Toolkit (InSTAnT)
Update in
-
Intracellular spatial transcriptomic analysis toolkit (InSTAnT).Nat Commun. 2024 Sep 6;15(1):7794. doi: 10.1038/s41467-024-49457-w. Nat Commun. 2024. PMID: 39242579 Free PMC article.
Abstract
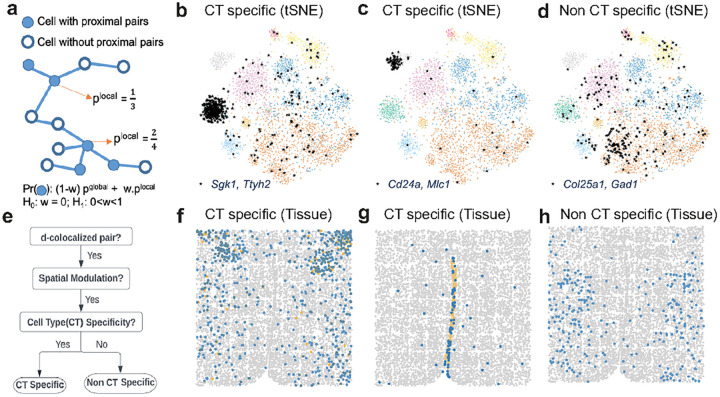
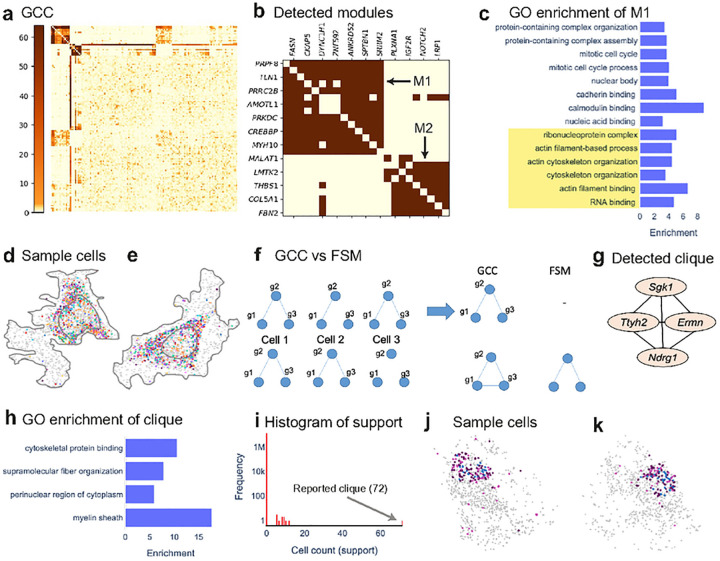
Imaging-based spatial transcriptomics technologies such as MERFISH offer snapshots of cellular processes in unprecedented detail, but new analytic tools are needed to realize their full potential. We present InSTAnT, a computational toolkit for extracting molecular relationships from spatial transcriptomics data at the intra-cellular resolution. InSTAnT detects gene pairs and modules with interesting patterns of mutual co-localization within and across cells, using specialized statistical tests and graph mining. We showcase the toolkit on datasets profiling a human cancer cell line and hypothalamic preoptic region of mouse brain. We performed rigorous statistical assessment of discovered co-localization patterns, found supporting evidence from databases and RNA interactions, and identified subcellular domains associated with RNA-colocalization. We identified several novel cell type-specific gene co-localizations in the brain. Intra-cellular spatial patterns discovered by InSTAnT mirror diverse molecular relationships, including RNA interactions and shared sub-cellular localization or function, providing a rich compendium of testable hypotheses regarding molecular functions.
Figures
References
-
- Marx V. Method of the Year: spatially resolved transcriptomics. Nature methods 18, 9–14 (2021). - PubMed
-
- Pham D. et al. stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. BioRxiv (2020).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
