

This is a preprint.
Human milk variation is shaped by maternal genetics and impacts the infant gut microbiome
- PMID: 36747843
- PMCID: PMC9900818
- DOI: 10.1101/2023.01.24.525211
Human milk variation is shaped by maternal genetics and impacts the infant gut microbiome
Update in
-
Human milk variation is shaped by maternal genetics and impacts the infant gut microbiome.Cell Genom. 2024 Oct 9;4(10):100638. doi: 10.1016/j.xgen.2024.100638. Epub 2024 Sep 11. Cell Genom. 2024. PMID: 39265573 Free PMC article.
Abstract
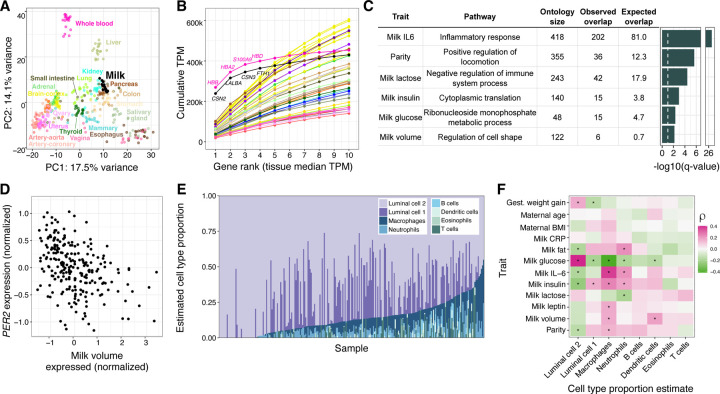
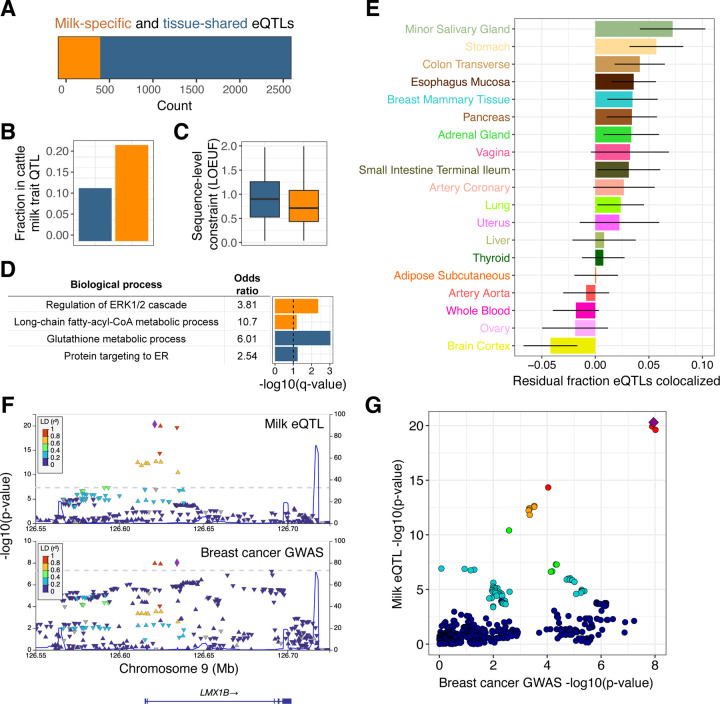
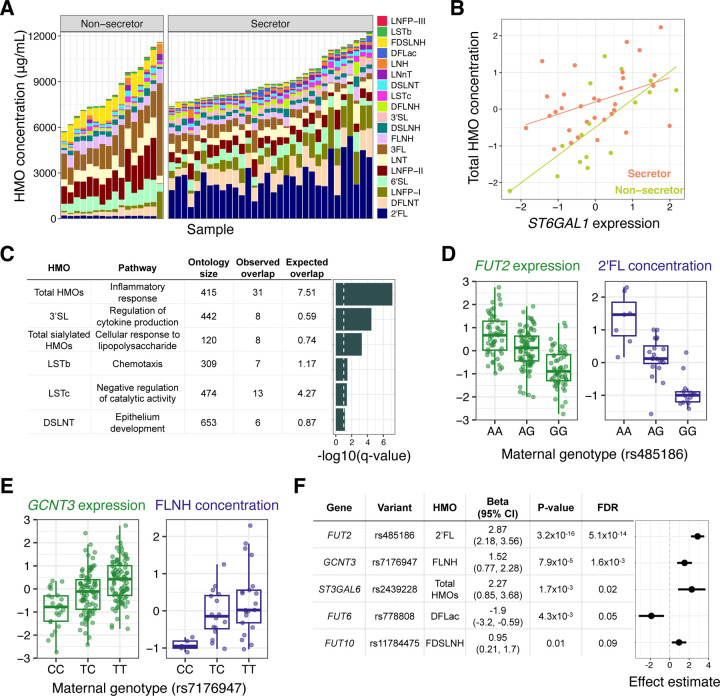
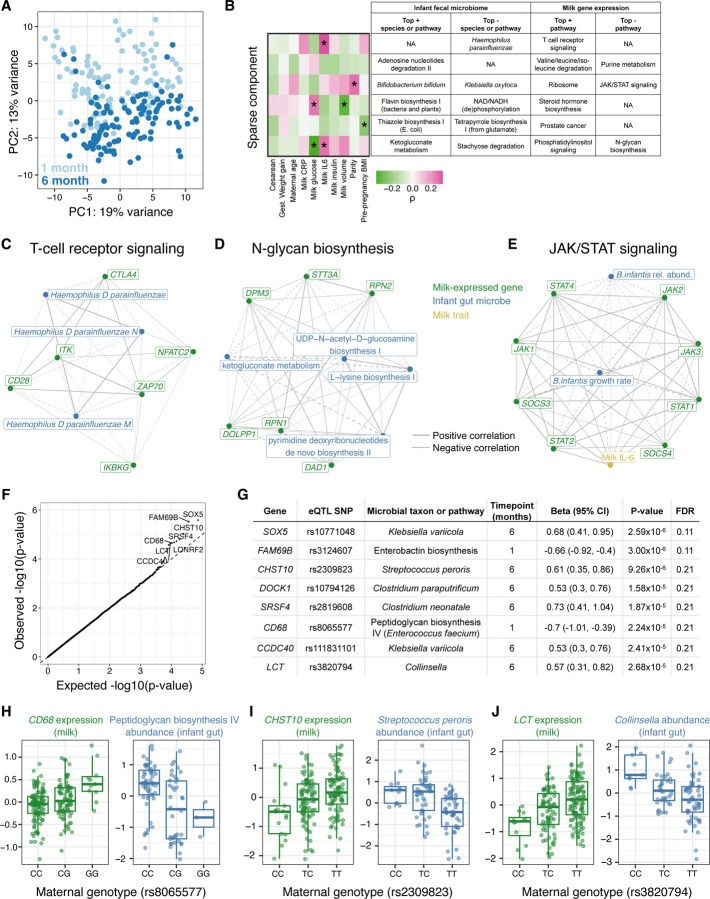
Human milk is a complex mix of nutritional and bioactive components that provide complete nutrition for the infant. However, we lack a systematic knowledge of the factors shaping milk composition and how milk variation influences infant health. Here, we used multi-omic profiling to characterize interactions between maternal genetics, milk gene expression, milk composition, and the infant fecal microbiome in 242 exclusively breastfeeding mother-infant pairs. We identified 487 genetic loci associated with milk gene expression unique to the lactating mammary gland, including loci that impacted breast cancer risk and human milk oligosaccharide concentration. Integrative analyses uncovered connections between milk gene expression and infant gut microbiome, including an association between the expression of inflammation-related genes with IL-6 concentration in milk and the abundance of Bifidobacteria in the infant gut. Our results show how an improved understanding of the genetics and genomics of human milk connects lactation biology with maternal and infant health.
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
-
- Lefèvre C. M., Sharp J. A. & Nicholas K. R. Evolution of lactation: ancient origin and extreme adaptations of the lactation system. Annu. Rev. Genomics Hum. Genet. 11, 219–238 (2010). - PubMed
-
- Victora C. G. et al. Breastfeeding in the 21st century: epidemiology, mechanisms, and lifelong effect. Lancet 387, 475–490 (2016). - PubMed
-
- Andreas N. J., Kampmann B. & Mehring Le-Doare K. Human breast milk: A review on its composition and bioactivity. Early Hum. Dev. 91, 629–635 (2015). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources