Multiplex MinION sequencing suggests enteric adenovirus F41 genetic diversity comparable to pre-COVID-19 era
- PMID: 36748435
- PMCID: PMC9973849
- DOI: 10.1099/mgen.0.000920
Multiplex MinION sequencing suggests enteric adenovirus F41 genetic diversity comparable to pre-COVID-19 era
Abstract
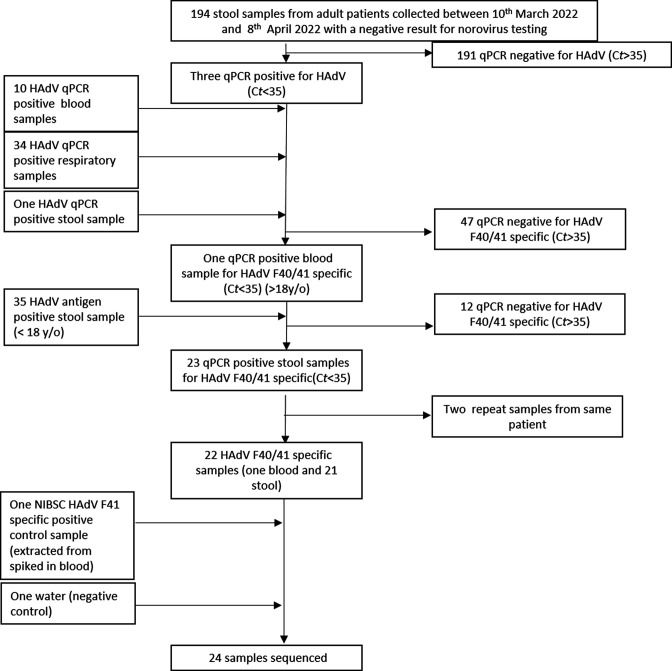
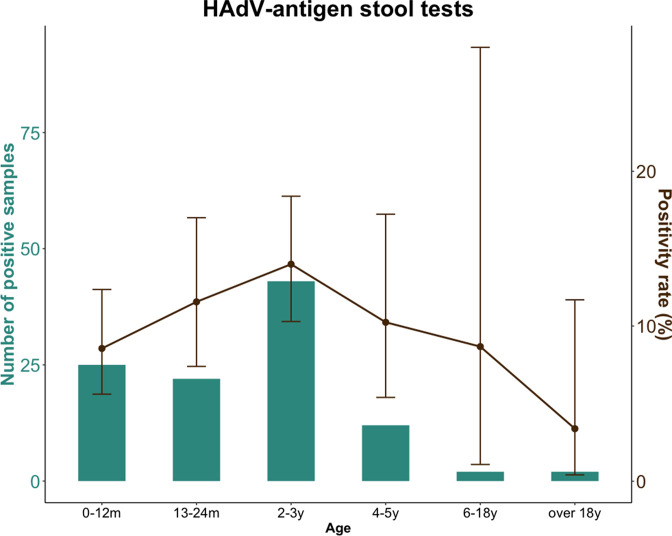
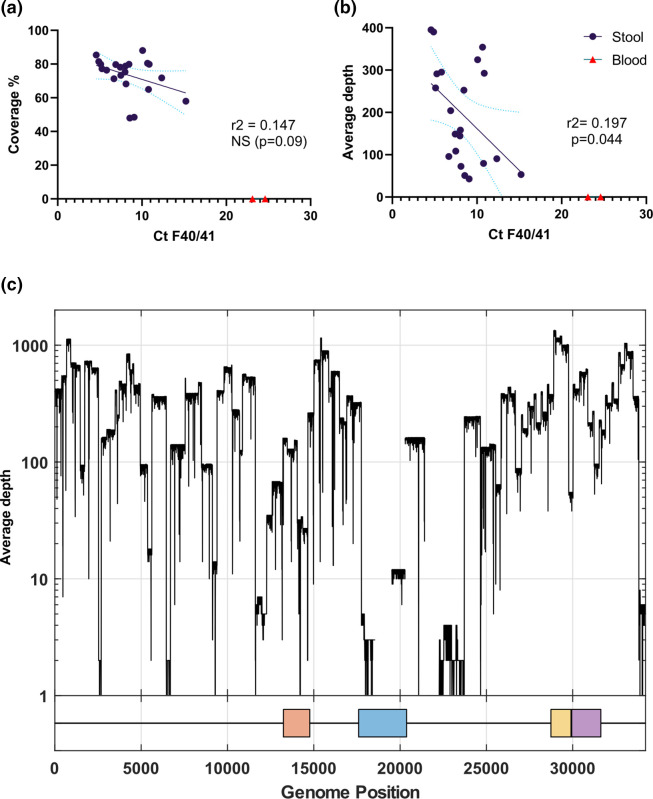
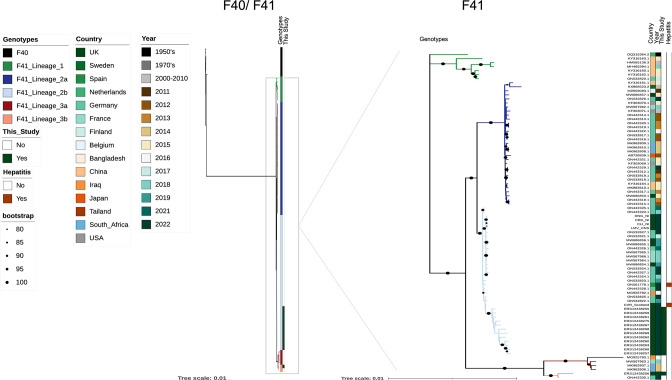
Human adenovirus F41 causes acute gastroenteritis in children, and has recently been associated with an apparent increase in paediatric hepatitis of unknown aetiology in the UK, with further cases reported in multiple countries. Relatively little is known about the genetic diversity of adenovirus F41 in UK children; and it is unclear what, if any, impact the COVID-19 pandemic has had on viral diversity in the UK. Methods that allow F41 to be sequenced from clinical samples without the need for viral culture are required to provide the genomic data to address these questions. Therefore, we evaluated an overlapping-amplicon method of sequencing adenovirus genomes from clinical samples using Oxford Nanopore technology. We applied this method to a small sample of adenovirus-species-F-positive extracts collected as part of standard care in the East of England region in January-May 2022. This method produced genomes with >75 % coverage in 13/22 samples and >50 % coverage in 19/22 samples. We identified two F41 lineages present in paediatric patients in the East of England in 2022. Where F41 genomes from paediatric hepatitis cases were available (n=2), these genomes fell within the diversity of F41 from the UK and continental Europe sequenced before and after the 2020-2021 phase of the COVID-19 pandemic. Our analyses suggest that overlapping amplicon sequencing is an appropriate method for generating F41 genomic data from high-virus-load clinical samples, and currently circulating F41 viral lineages were present in the UK and Europe before the COVID-19 pandemic.
Keywords: DNA virus; adenovirus; genetic epidemiology; genomics; hepatitis; virology.
Conflict of interest statement
The authors declare that there are no conflicts of interest
Figures