

Genetic determinants of host tropism in Klebsiella phages
- PMID: 36753420
- PMCID: PMC9989827
- DOI: 10.1016/j.celrep.2023.112048
Genetic determinants of host tropism in Klebsiella phages
Abstract
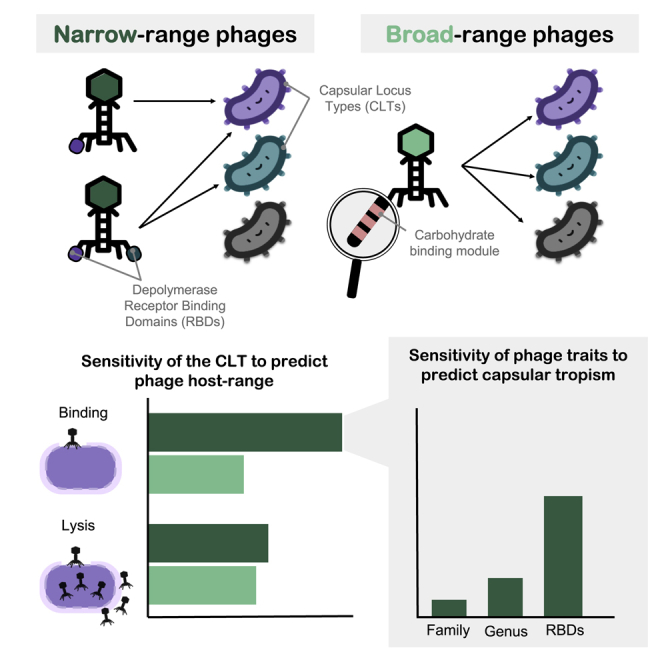
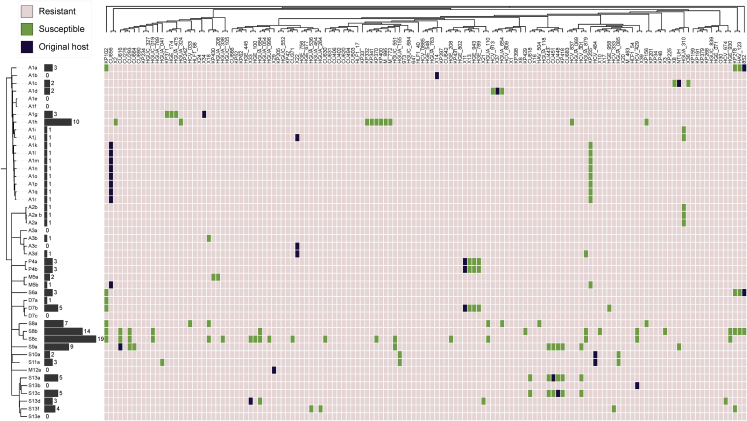
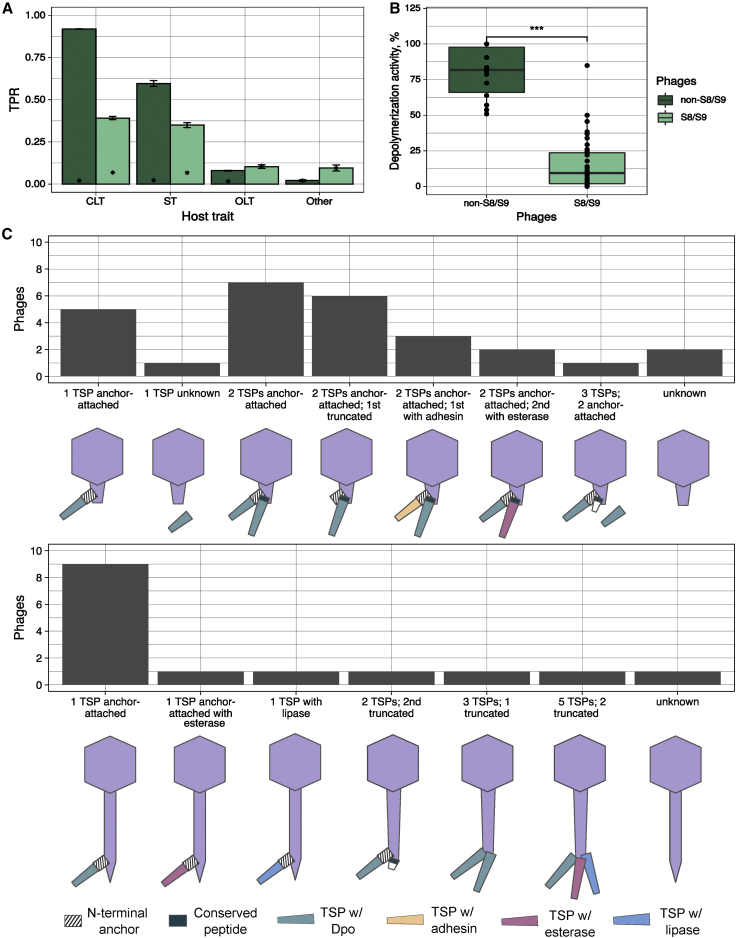
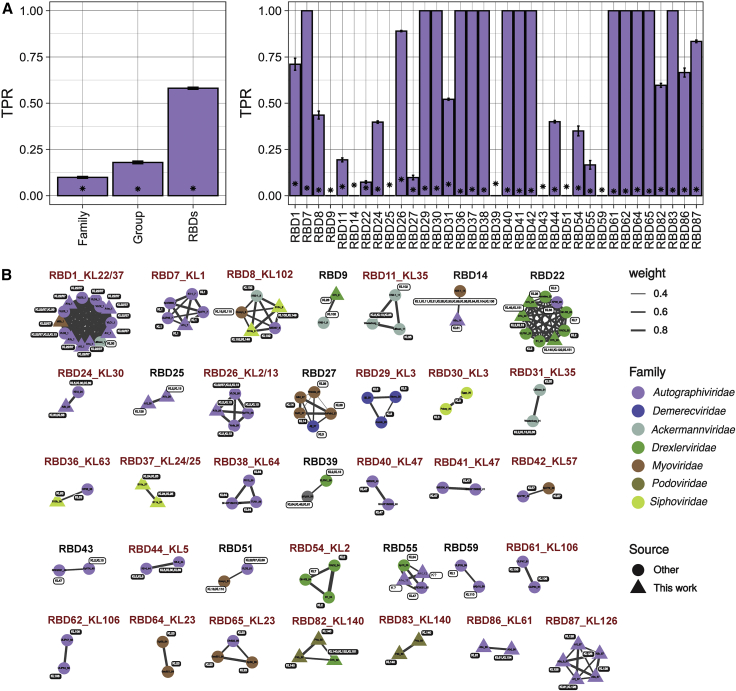
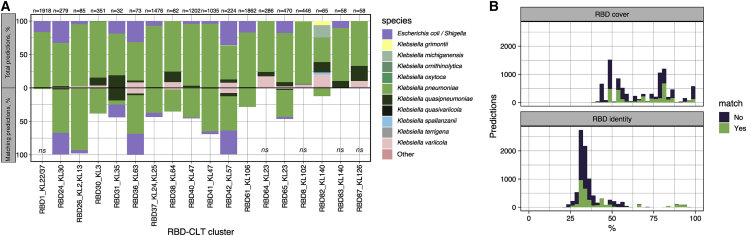
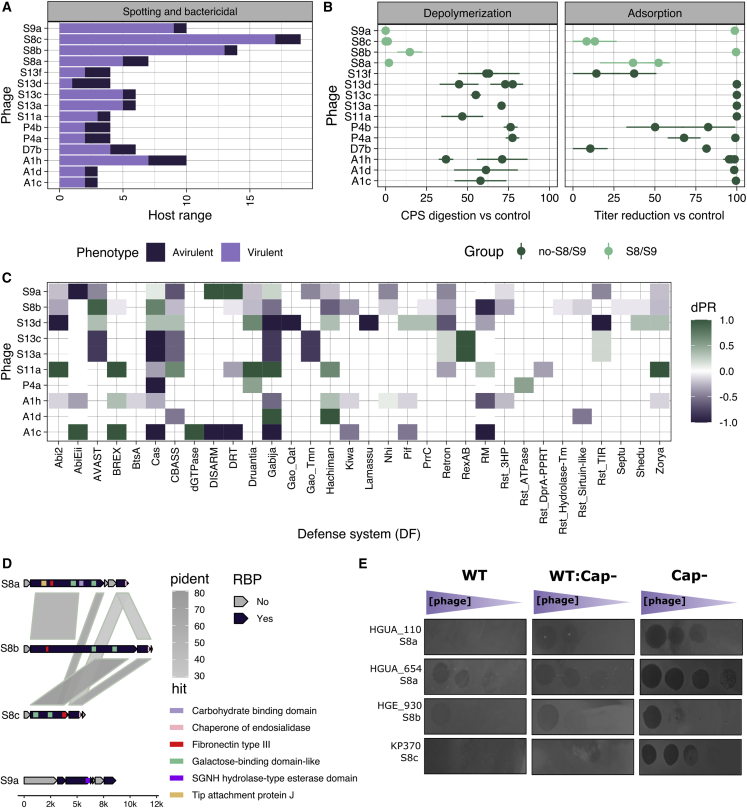
Bacteriophages play key roles in bacterial ecology and evolution and are potential antimicrobials. However, the determinants of phage-host specificity remain elusive. Here, we isolate 46 phages to challenge 138 representative clinical isolates of Klebsiella pneumoniae, a widespread opportunistic pathogen. Spot tests show a narrow host range for most phages, with <2% of 6,319 phage-host combinations tested yielding detectable interactions. Bacterial capsule diversity is the main factor restricting phage host range. Consequently, phage-encoded depolymerases are key determinants of host tropism, and depolymerase sequence types are associated with the ability to infect specific capsular types across phage families. However, all phages with a broader host range found do not encode canonical depolymerases, suggesting alternative modes of entry. These findings expand our knowledge of the complex interactions between bacteria and their viruses and point out the feasibility of predicting the first steps of phage infection using bacterial and phage genome sequences.
Keywords: CP: Microbiology; Klebsiella; bacterial capsule; bacteriophage; depolymerase; genomics; horizontal gene transfer; host range; microbial evolution.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
-
- Abedon S.T. Cambridge University Press; 2008. Bacteriophage Ecology: Population Growth, Evolution, and Impact of Bacterial Viruses.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
