

Single-cell profiling of alveolar rhabdomyosarcoma reveals RAS pathway inhibitors as cell-fate hijackers with therapeutic relevance
- PMID: 36753540
- PMCID: PMC9908029
- DOI: 10.1126/sciadv.ade9238
Single-cell profiling of alveolar rhabdomyosarcoma reveals RAS pathway inhibitors as cell-fate hijackers with therapeutic relevance
Erratum in
-
Erratum for the Research Article "Single-cell profiling of alveolar rhabdomyosarcoma reveals RAS pathway inhibitors as cell-fate hijackers with therapeutic relevance" by S. G. Danielli et al.Sci Adv. 2025 Dec 12;11(50):eaee1296. doi: 10.1126/sciadv.aee1296. Epub 2025 Dec 12. Sci Adv. 2025. PMID: 41385647 Free PMC article. No abstract available.
Abstract
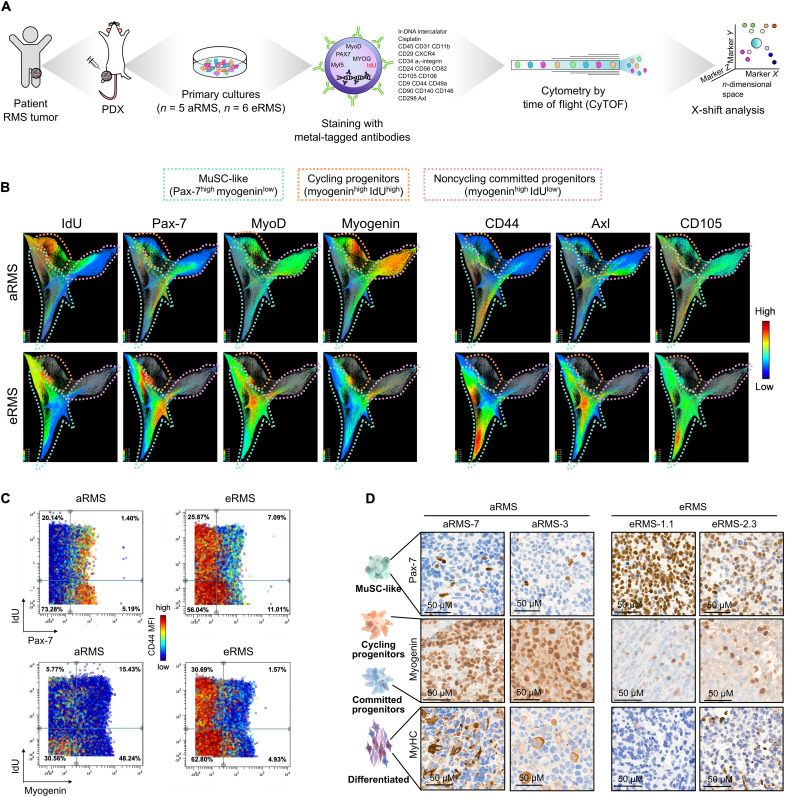
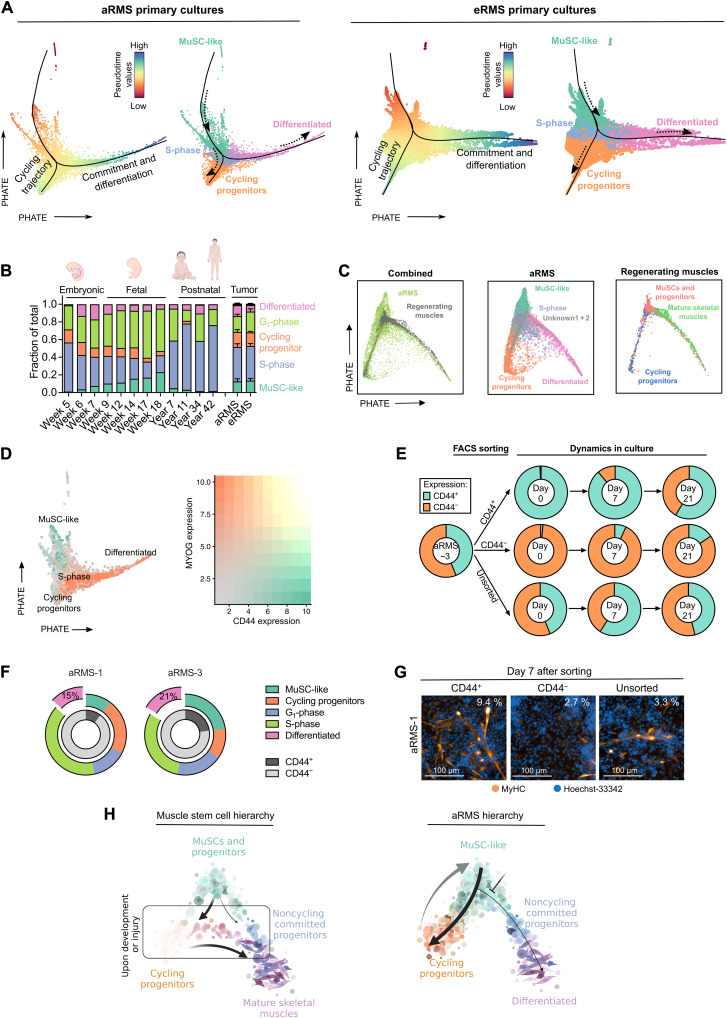
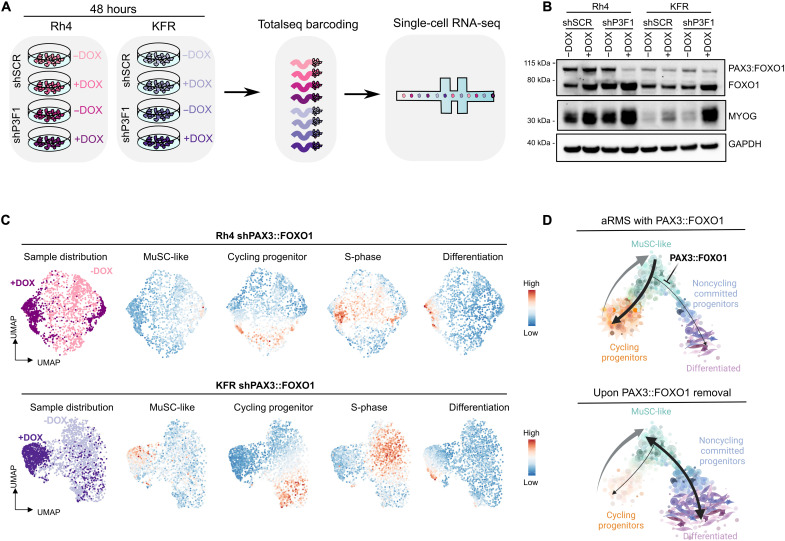
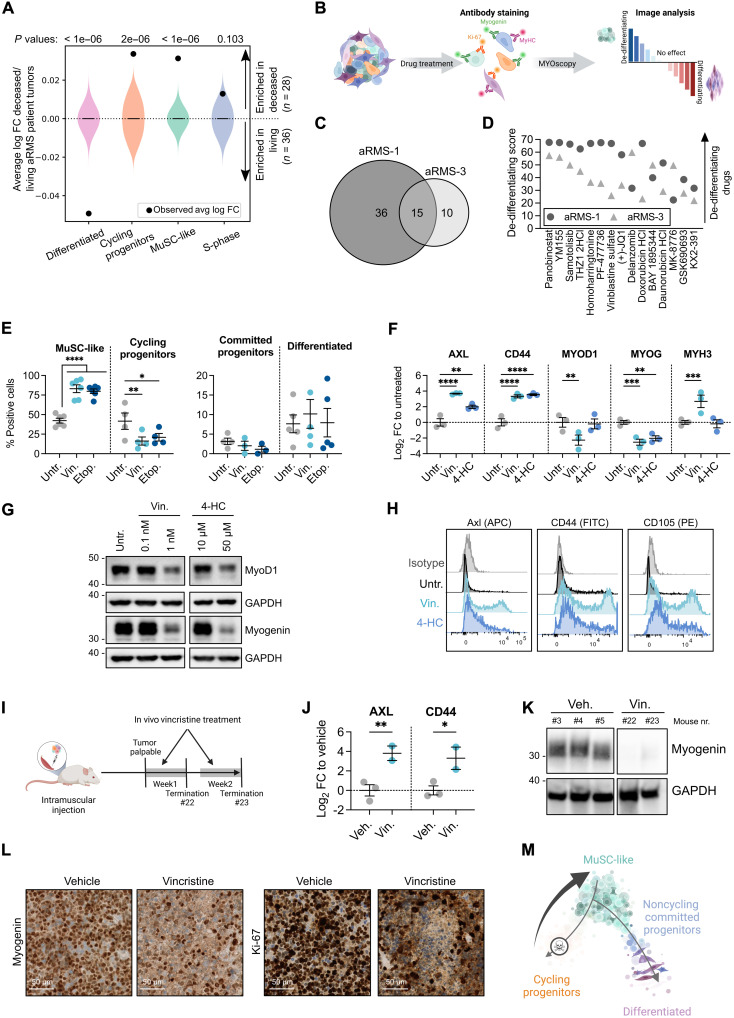
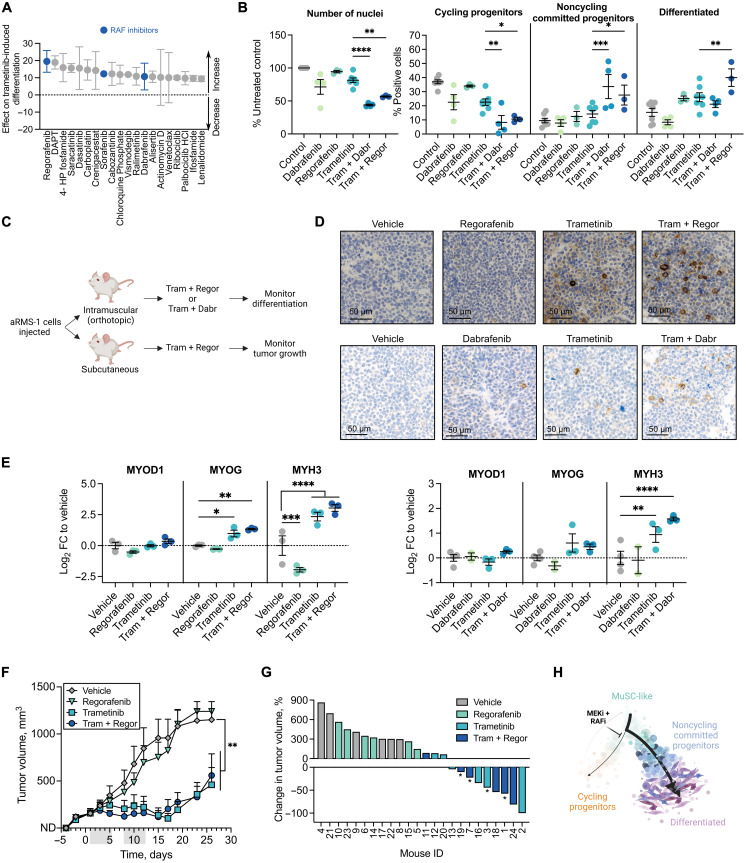
Rhabdomyosarcoma (RMS) is a group of pediatric cancers with features of developing skeletal muscle. The cellular hierarchy and mechanisms leading to developmental arrest remain elusive. Here, we combined single-cell RNA sequencing, mass cytometry, and high-content imaging to resolve intratumoral heterogeneity of patient-derived primary RMS cultures. We show that the aggressive alveolar RMS (aRMS) subtype contains plastic muscle stem-like cells and cycling progenitors that drive tumor growth, and a subpopulation of differentiated cells that lost its proliferative potential and correlates with better outcomes. While chemotherapy eliminates cycling progenitors, it enriches aRMS for muscle stem-like cells. We screened for drugs hijacking aRMS toward clinically favorable subpopulations and identified a combination of RAF and MEK inhibitors that potently induces myogenic differentiation and inhibits tumor growth. Overall, our work provides insights into the developmental states underlying aRMS aggressiveness, chemoresistance, and progression and identifies the RAS pathway as a promising therapeutic target.
Figures


References
-
- Coorens T. H. H., Behjati S., Tracing and targeting the origins of childhood cancer. Annu. Rev. Cancer Biol. 6, 35–47 (2022).
-
- Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020). - PubMed
-
- Hanahan D., Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46 (2022). - PubMed
-
- Jessa S., Blanchet-Cohen A., Krug B., Vladoiu M., Coutelier M., Faury D., Poreau B., De Jay N., Hébert S., Monlong J., Farmer W. T., Donovan L. K., Hu Y., McConechy M. K., Cavalli F. M. G., Mikael L. G., Ellezam B., Richer M., Allaire A., Weil A. G., Atkinson J., Farmer J.-P., Dudley R. W. R., Larouche V., Crevier L., Albrecht S., Filbin M. G., Sartelet H., Lutz P.-E., Nagy C., Turecki G., Costantino S., Dirks P. B., Murai K. K., Bourque G., Ragoussis J., Garzia L., Taylor M. D., Jabado N., Kleinman C. L., Stalled developmental programs at the root of pediatric brain tumors. Nat. Genet. 51, 1702–1713 (2019). - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
