

A Clinical Workflow for Cost-Saving High-Rate Diagnosis of Genetic Kidney Diseases
- PMID: 36753701
- PMCID: PMC10103218
- DOI: 10.1681/ASN.0000000000000076
A Clinical Workflow for Cost-Saving High-Rate Diagnosis of Genetic Kidney Diseases
Abstract
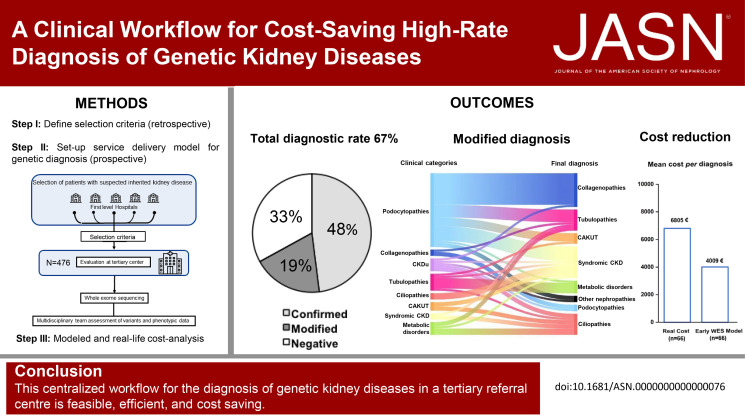
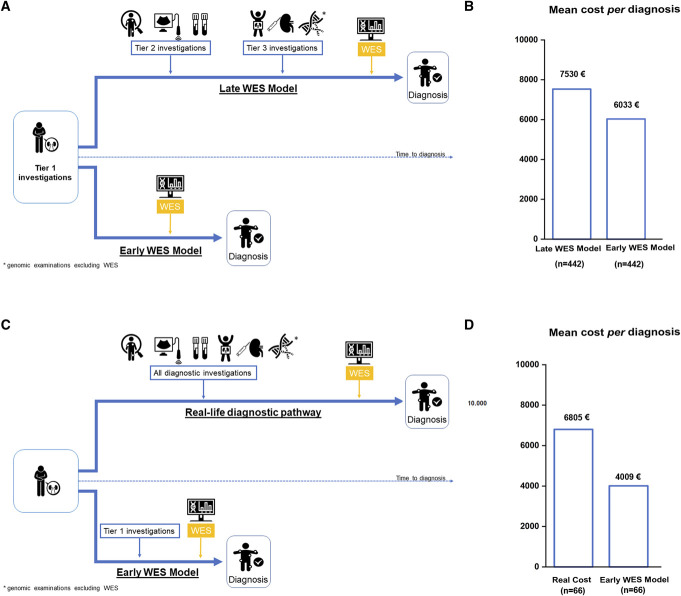
Significance statement: To optimize the diagnosis of genetic kidney disorders in a cost-effective manner, we developed a workflow based on referral criteria for in-person evaluation at a tertiary center, whole-exome sequencing, reverse phenotyping, and multidisciplinary board analysis. This workflow reached a diagnostic rate of 67%, with 48% confirming and 19% modifying the suspected clinical diagnosis. We obtained a genetic diagnosis in 64% of children and 70% of adults. A modeled cost analysis demonstrated that early genetic testing saves 20% of costs per patient. Real cost analysis on a representative sample of 66 patients demonstrated an actual cost reduction of 41%. This workflow demonstrates feasibility, performance, and economic effect for the diagnosis of genetic kidney diseases in a real-world setting.
Background: Whole-exome sequencing (WES) increases the diagnostic rate of genetic kidney disorders, but accessibility, interpretation of results, and costs limit use in daily practice.
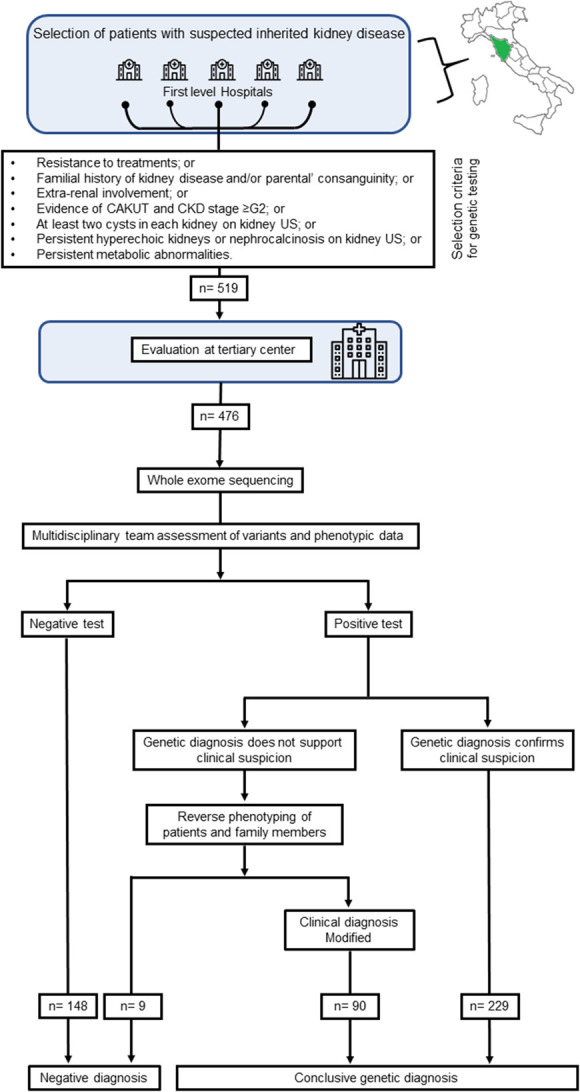
Methods: Univariable analysis of a historical cohort of 392 patients who underwent WES for kidney diseases showed that resistance to treatments, familial history of kidney disease, extrarenal involvement, congenital abnormalities of the kidney and urinary tract and CKD stage ≥G2, two or more cysts per kidney on ultrasound, persistent hyperechoic kidneys or nephrocalcinosis on ultrasound, and persistent metabolic abnormalities were most predictive for genetic diagnosis. We prospectively applied these criteria to select patients in a network of nephrology centers, followed by centralized genetic diagnosis by WES, reverse phenotyping, and multidisciplinary board discussion.
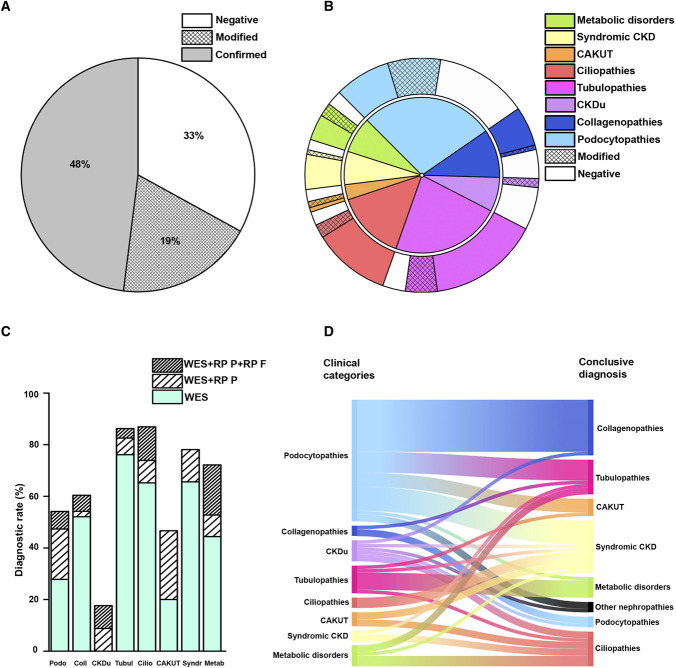
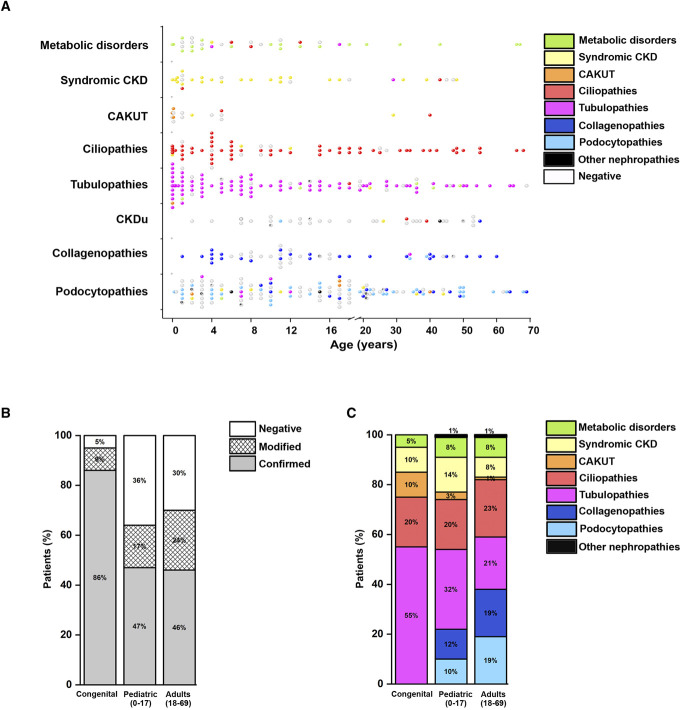
Results: We applied this multistep workflow to 476 patients with eight clinical categories (podocytopathies, collagenopathies, CKD of unknown origin, tubulopathies, ciliopathies, congenital anomalies of the kidney and urinary tract, syndromic CKD, metabolic kidney disorders), obtaining genetic diagnosis for 319 of 476 patients (67.0%) (95% in 21 patients with disease onset during the fetal period or at birth, 64% in 298 pediatric patients, and 70% in 156 adult patients). The suspected clinical diagnosis was confirmed in 48% of the 476 patients and modified in 19%. A modeled cost analysis showed that application of this workflow saved 20% of costs per patient when performed at the beginning of the diagnostic process. Real cost analysis of 66 patients randomly selected from all categories showed actual cost reduction of 41%.
Conclusions: A diagnostic workflow for genetic kidney diseases that includes WES is cost-saving, especially if implemented early, and is feasible in a real-world setting.
Copyright © 2023 by the American Society of Nephrology.
Figures
Comment in
-
Kidney Genetics: Continuing Discoveries and a Roadmap to the Clinic.J Am Soc Nephrol. 2023 Apr 1;34(4):519-520. doi: 10.1681/ASN.0000000000000077. Epub 2023 Feb 9. J Am Soc Nephrol. 2023. PMID: 36758119 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
