

AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms
- PMID: 36755055
- PMCID: PMC9908985
- DOI: 10.1038/s42003-023-04488-9
AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms
Abstract
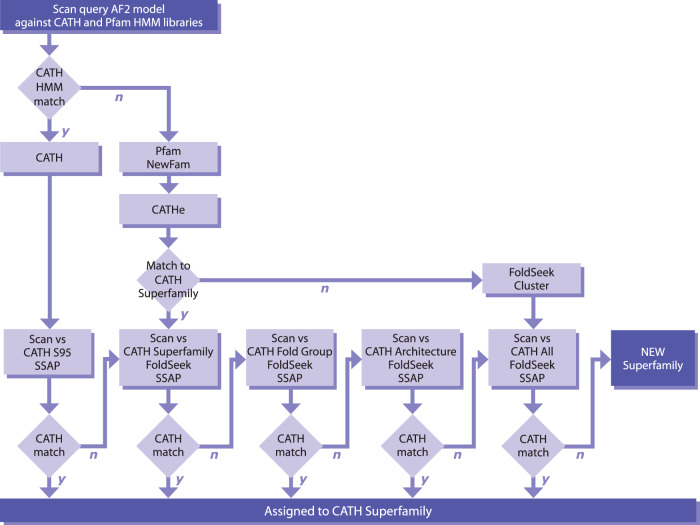
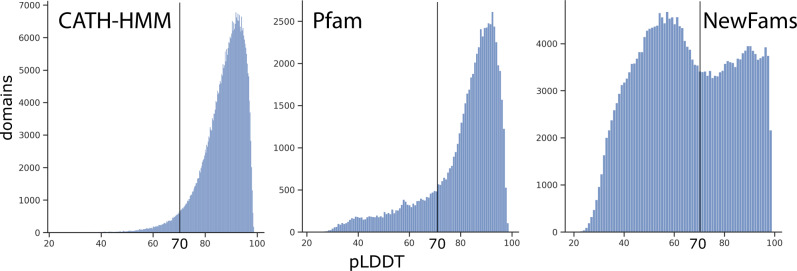
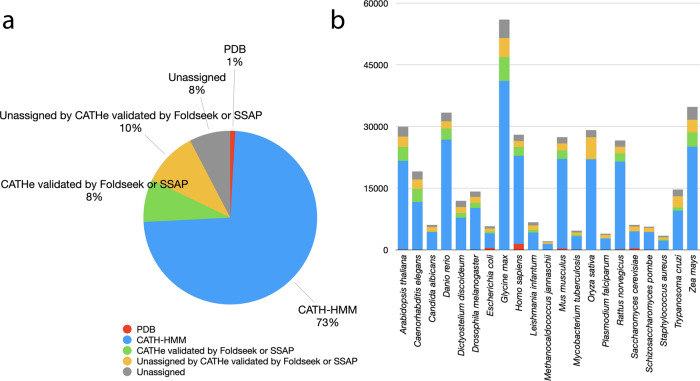
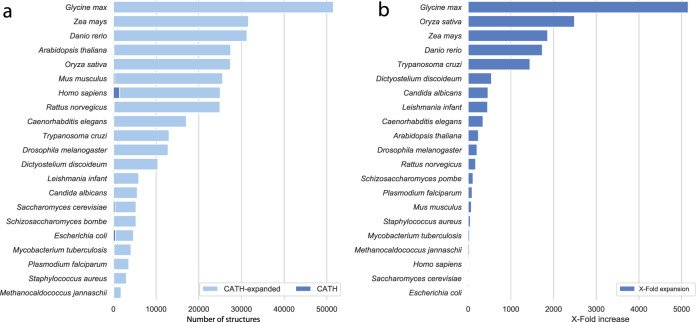
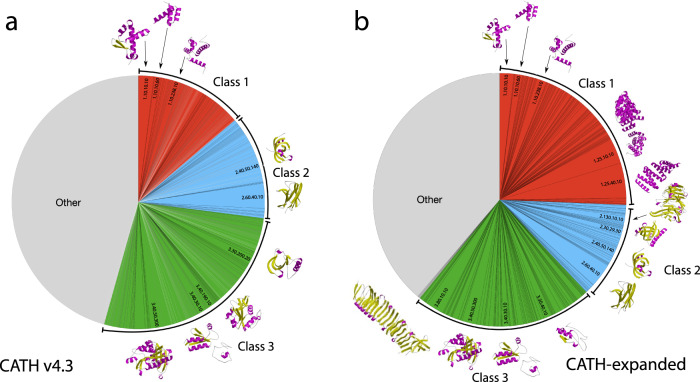
Deep-learning (DL) methods like DeepMind's AlphaFold2 (AF2) have led to substantial improvements in protein structure prediction. We analyse confident AF2 models from 21 model organisms using a new classification protocol (CATH-Assign) which exploits novel DL methods for structural comparison and classification. Of ~370,000 confident models, 92% can be assigned to 3253 superfamilies in our CATH domain superfamily classification. The remaining cluster into 2367 putative novel superfamilies. Detailed manual analysis on 618 of these, having at least one human relative, reveal extremely remote homologies and further unusual features. Only 25 novel superfamilies could be confirmed. Although most models map to existing superfamilies, AF2 domains expand CATH by 67% and increases the number of unique 'global' folds by 36% and will provide valuable insights on structure function relationships. CATH-Assign will harness the huge expansion in structural data provided by DeepMind to rationalise evolutionary changes driving functional divergence.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
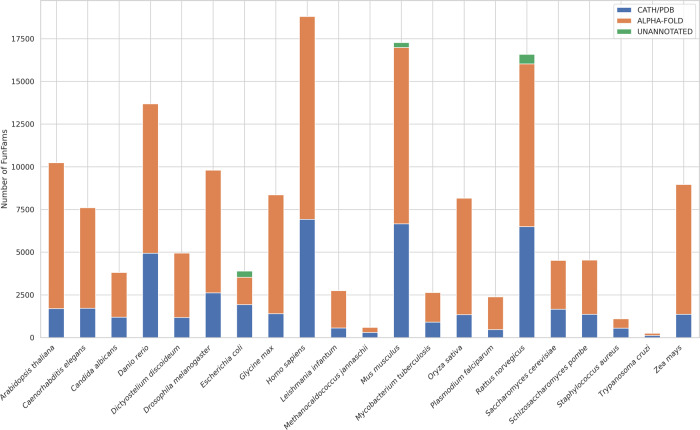
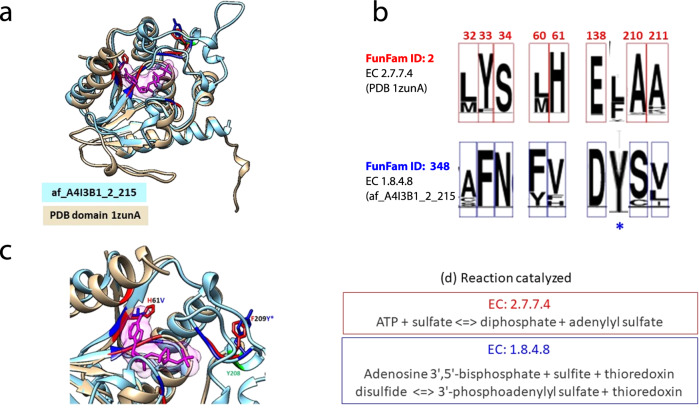
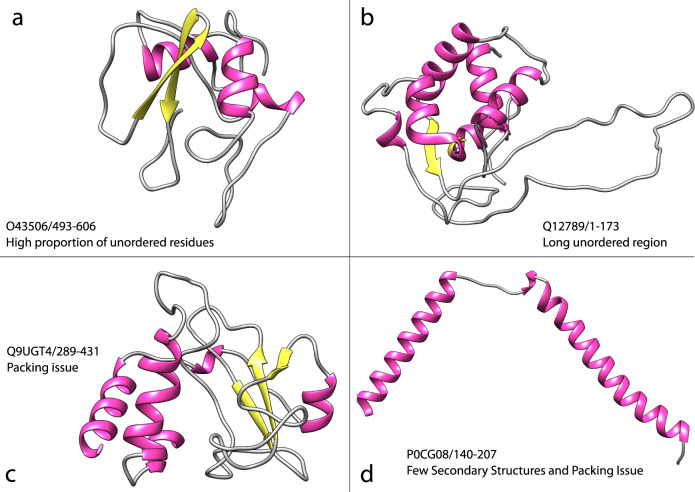
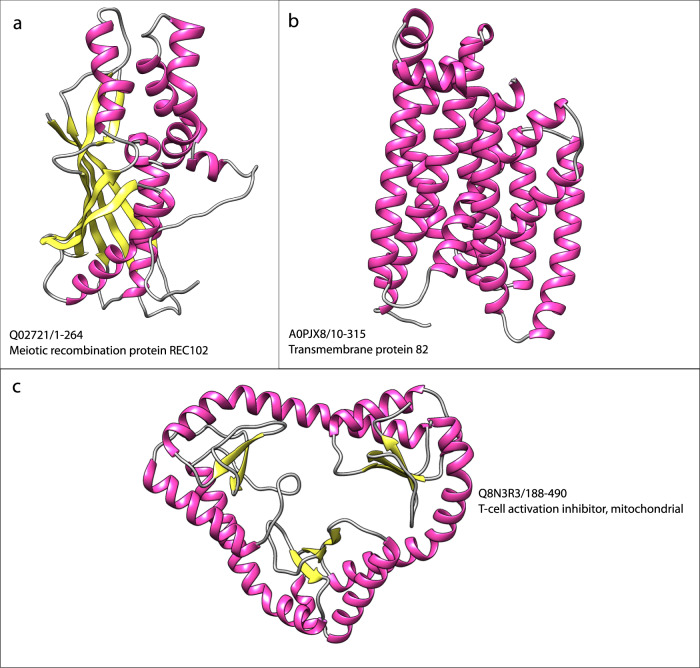
Comment in
-
Predicted protein structures expand the CATH database.Nat Methods. 2023 Apr;20(4):483. doi: 10.1038/s41592-023-01857-4. Nat Methods. 2023. PMID: 37046018 No abstract available.
References
-
- Gromiha, M. M., Nagarajan, R. & Selvaraj, S. Encyclopedia of Bioinformatics and Computational Biology 445–459 (Elsevier, 2019).
Publication types
MeSH terms
Substances
Grants and funding
- 221327/Z/20/Z/WT_/Wellcome Trust/United Kingdom
- BB/R009597/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/S020144/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/V014722/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/R014892/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
