

Dissecting cell identity via network inference and in silico gene perturbation
- PMID: 36755098
- PMCID: PMC9946838
- DOI: 10.1038/s41586-022-05688-9
Dissecting cell identity via network inference and in silico gene perturbation
Abstract
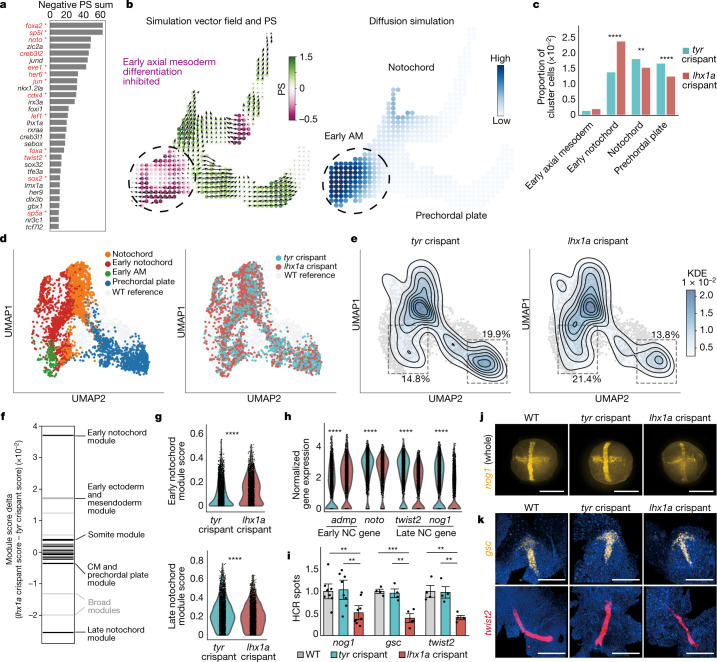
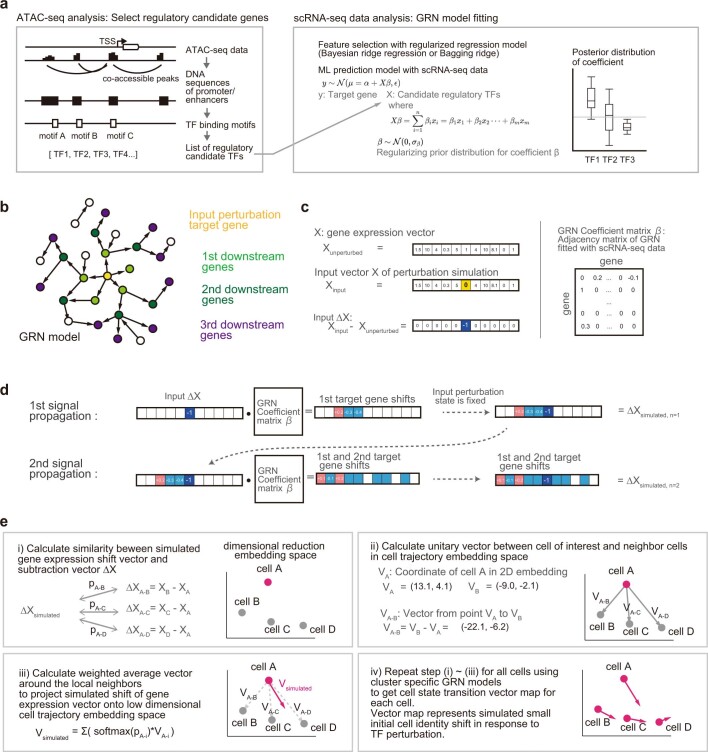
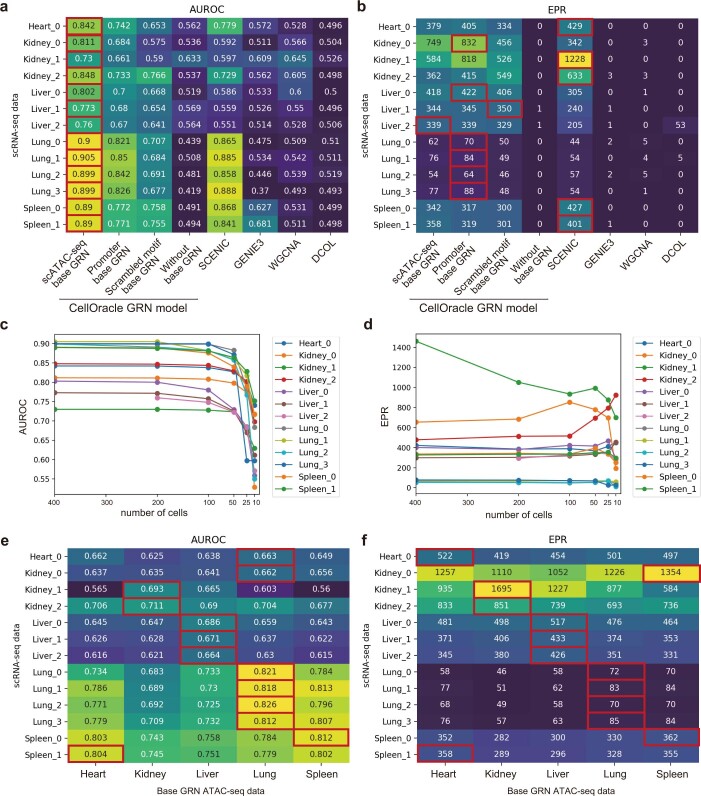
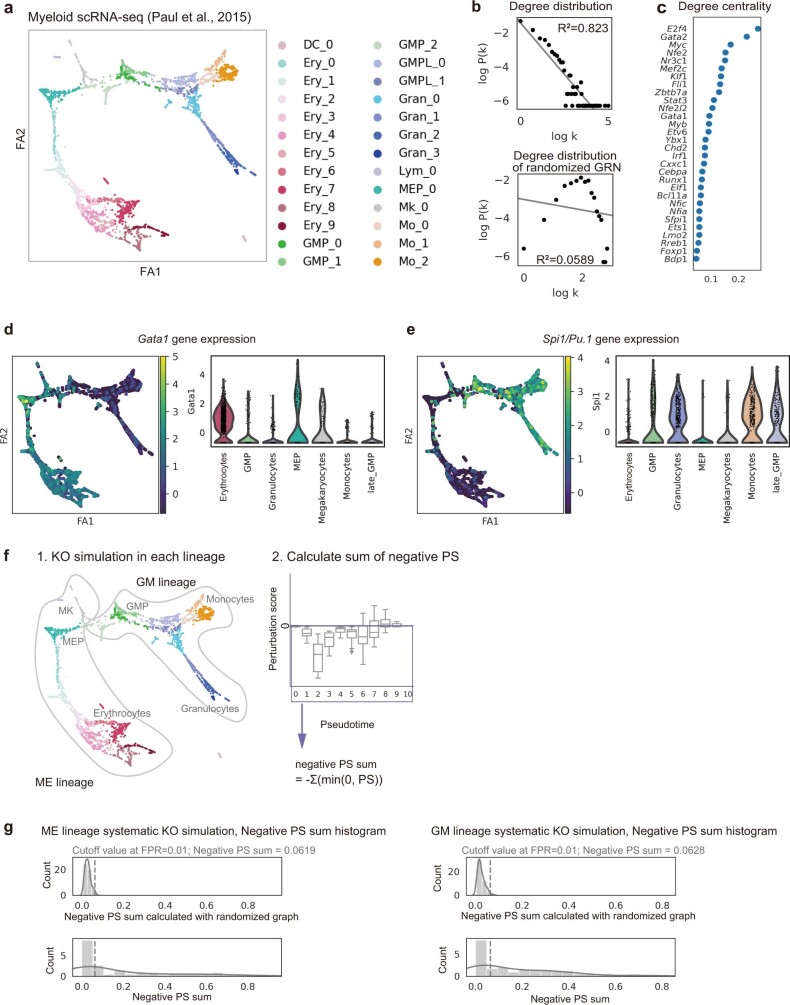
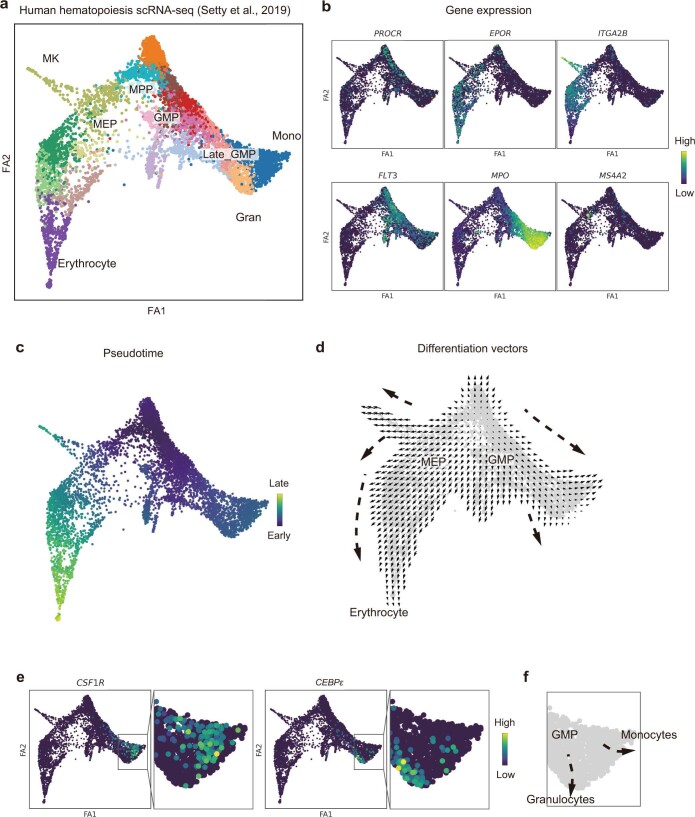
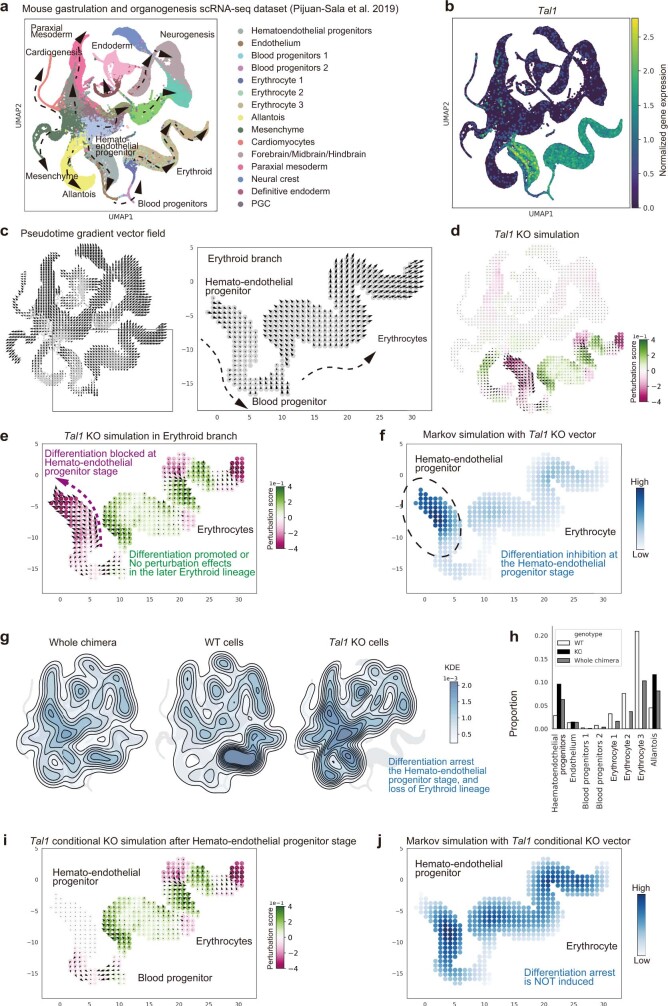
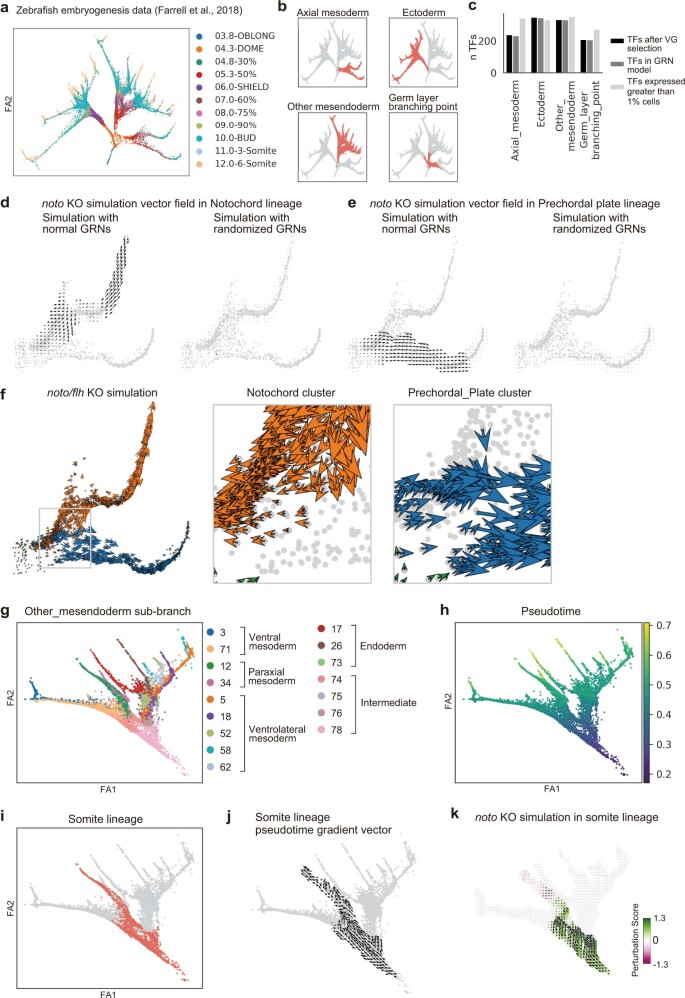
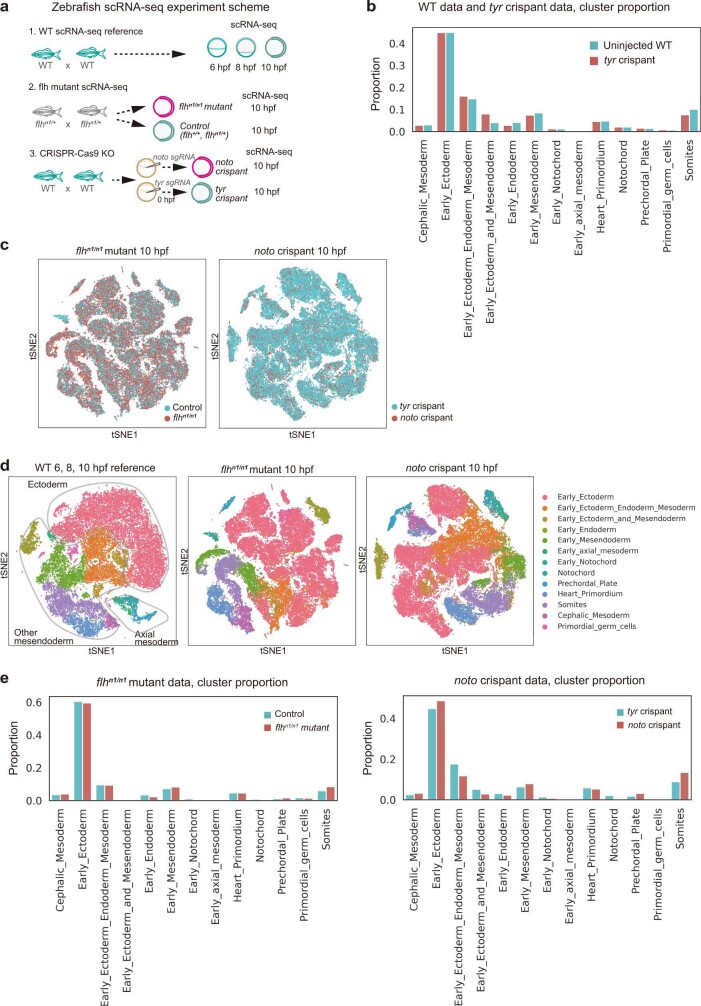
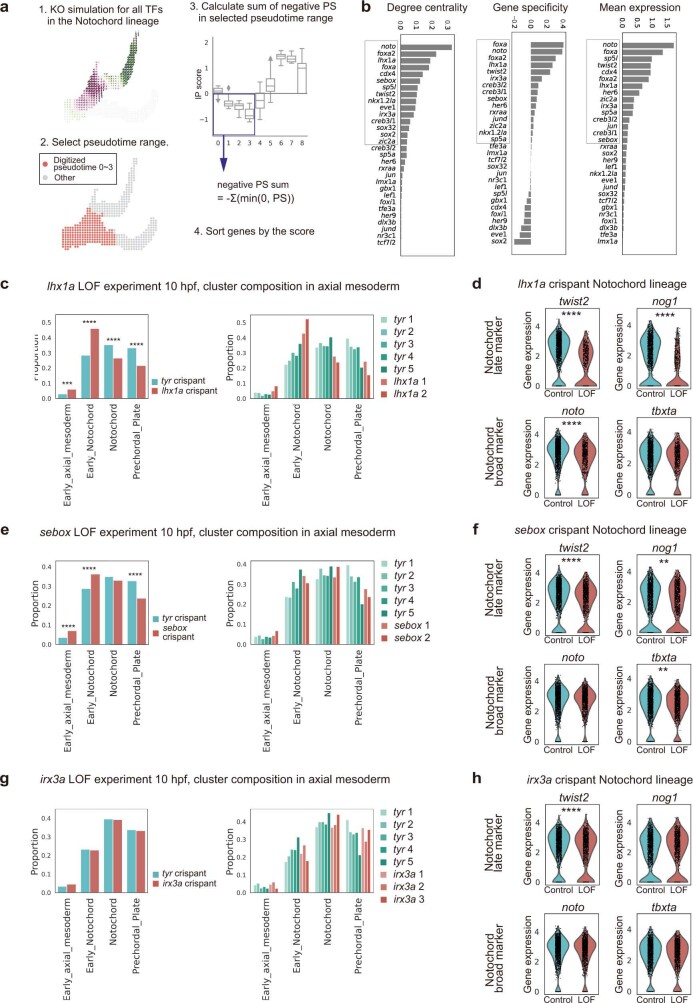
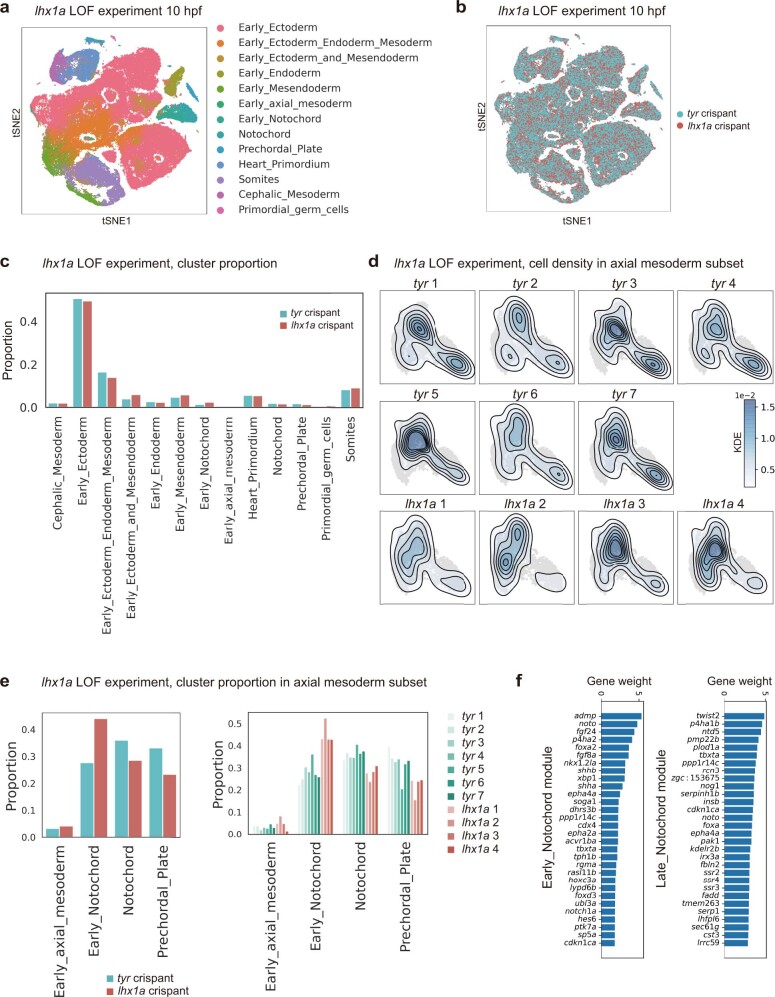
Cell identity is governed by the complex regulation of gene expression, represented as gene-regulatory networks1. Here we use gene-regulatory networks inferred from single-cell multi-omics data to perform in silico transcription factor perturbations, simulating the consequent changes in cell identity using only unperturbed wild-type data. We apply this machine-learning-based approach, CellOracle, to well-established paradigms-mouse and human haematopoiesis, and zebrafish embryogenesis-and we correctly model reported changes in phenotype that occur as a result of transcription factor perturbation. Through systematic in silico transcription factor perturbation in the developing zebrafish, we simulate and experimentally validate a previously unreported phenotype that results from the loss of noto, an established notochord regulator. Furthermore, we identify an axial mesoderm regulator, lhx1a. Together, these results show that CellOracle can be used to analyse the regulation of cell identity by transcription factors, and can provide mechanistic insights into development and differentiation.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
An oracle predicts regulators of cell identity.Nature. 2023 Feb;614(7949):630-632. doi: 10.1038/d41586-023-00251-6. Nature. 2023. PMID: 36755144 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
