

Regulation of DNA damage response by trimeric G-proteins
- PMID: 36756378
- PMCID: PMC9900518
- DOI: 10.1016/j.isci.2023.105973
Regulation of DNA damage response by trimeric G-proteins
Abstract
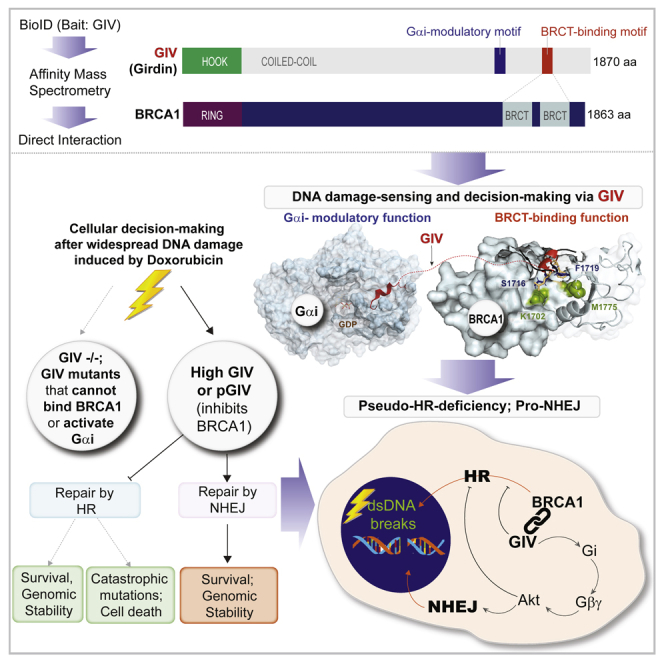
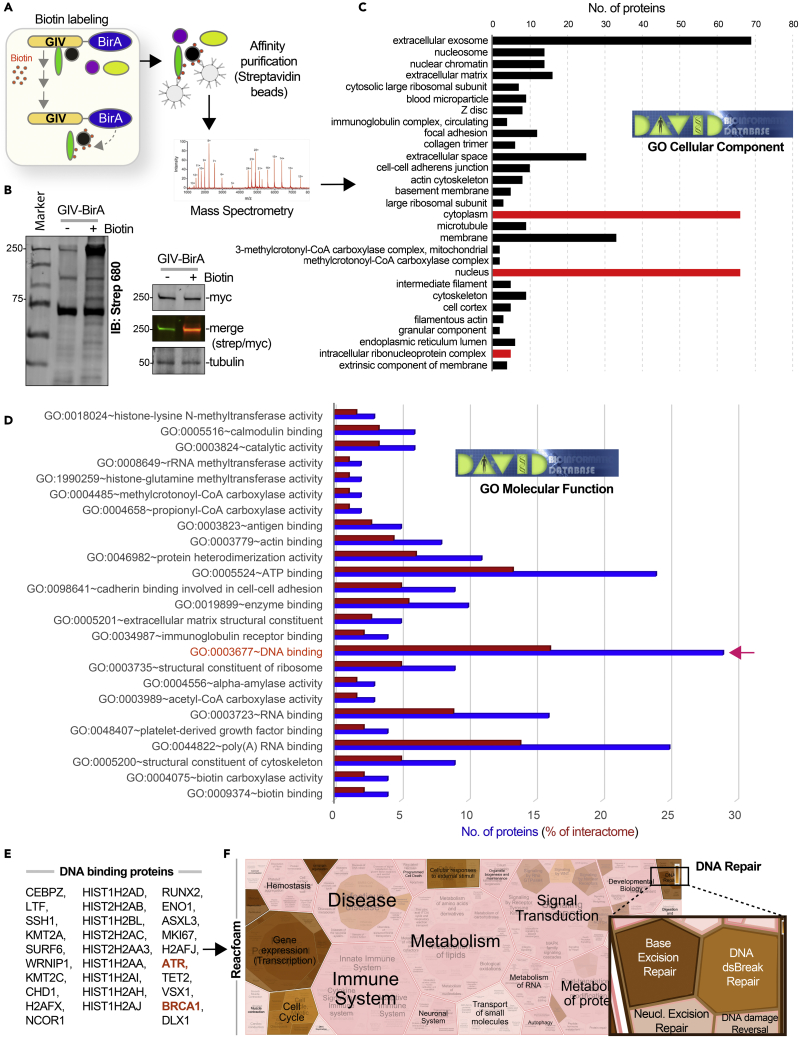
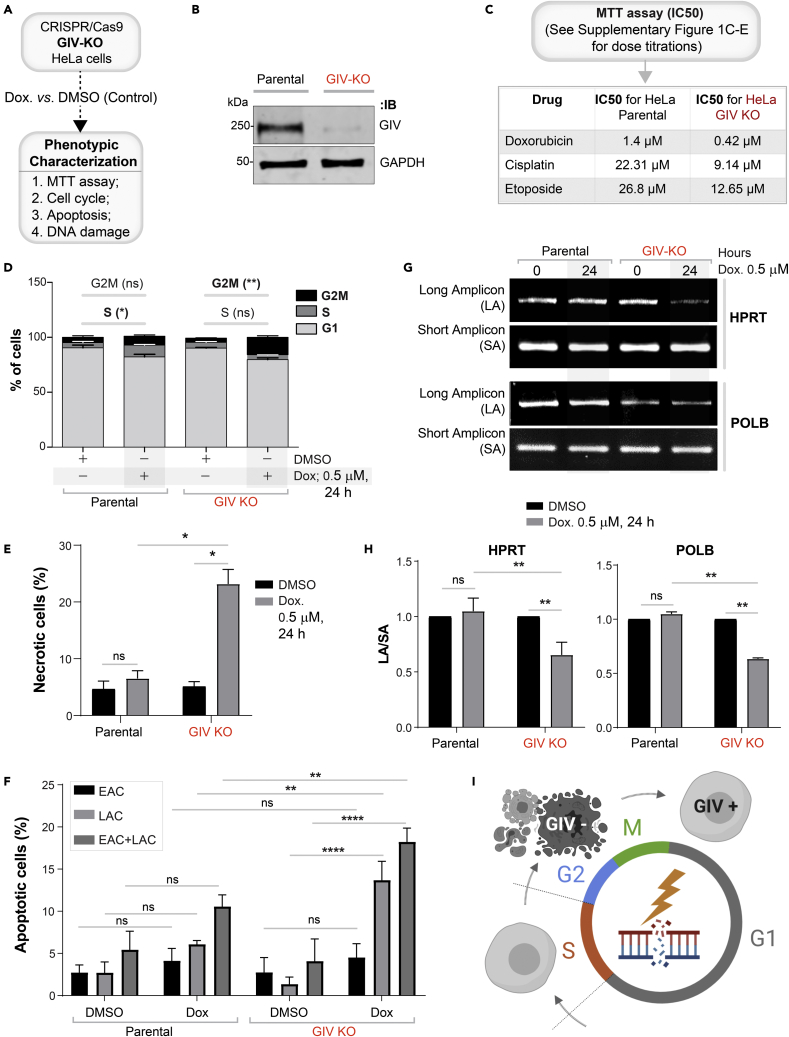
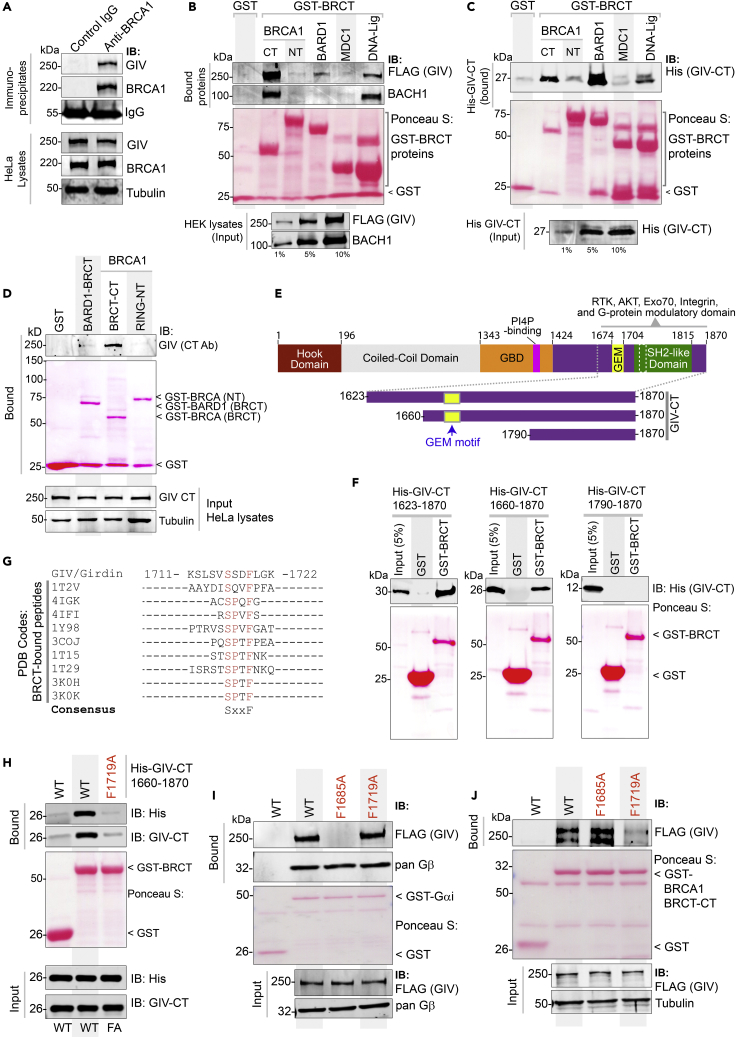
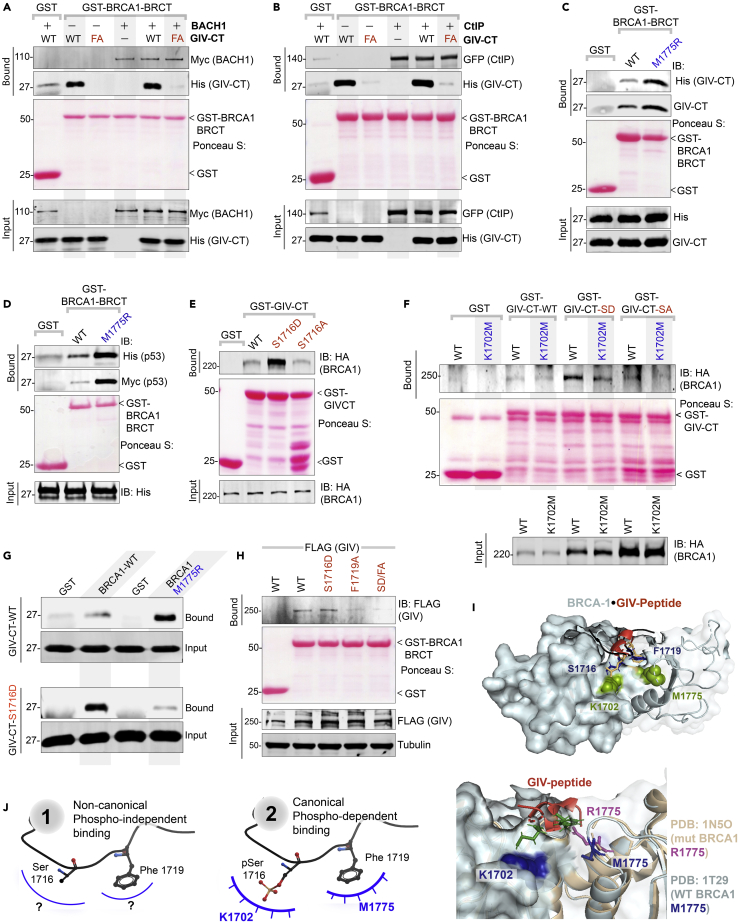
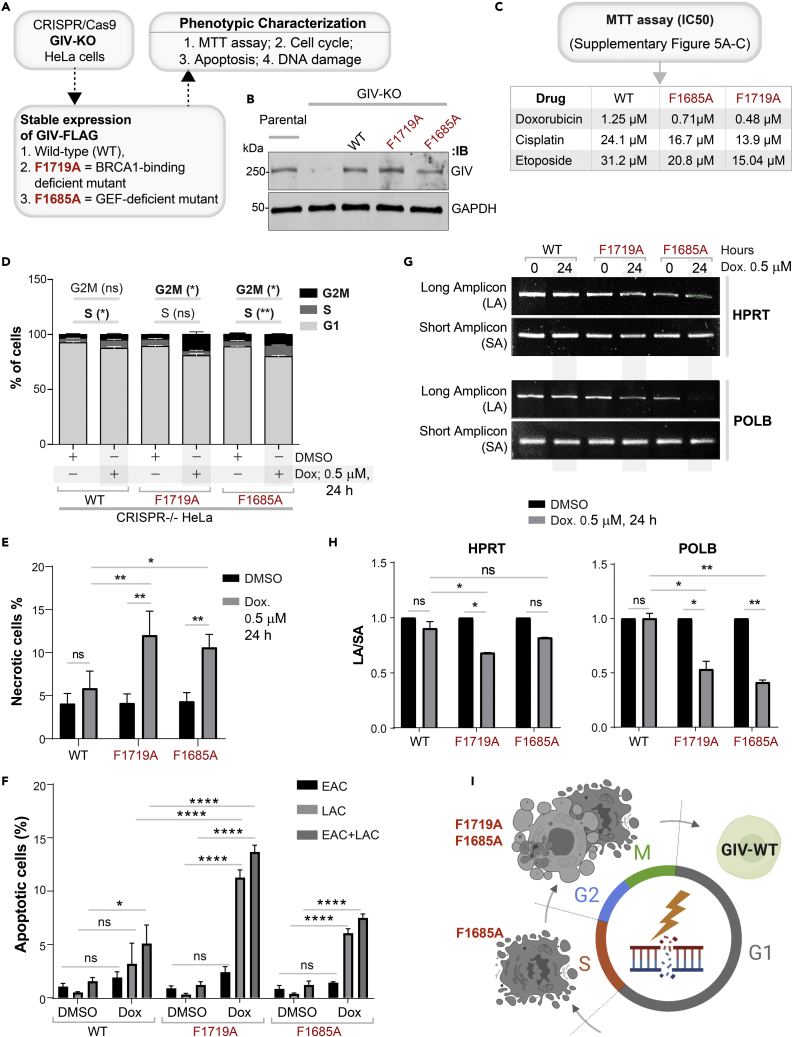
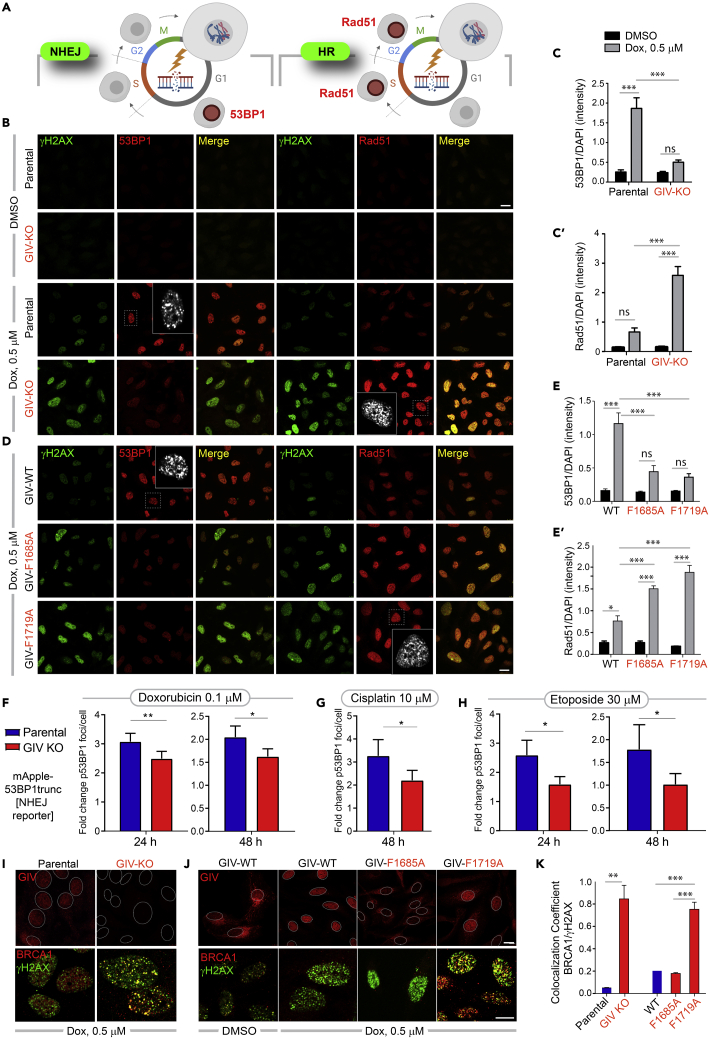
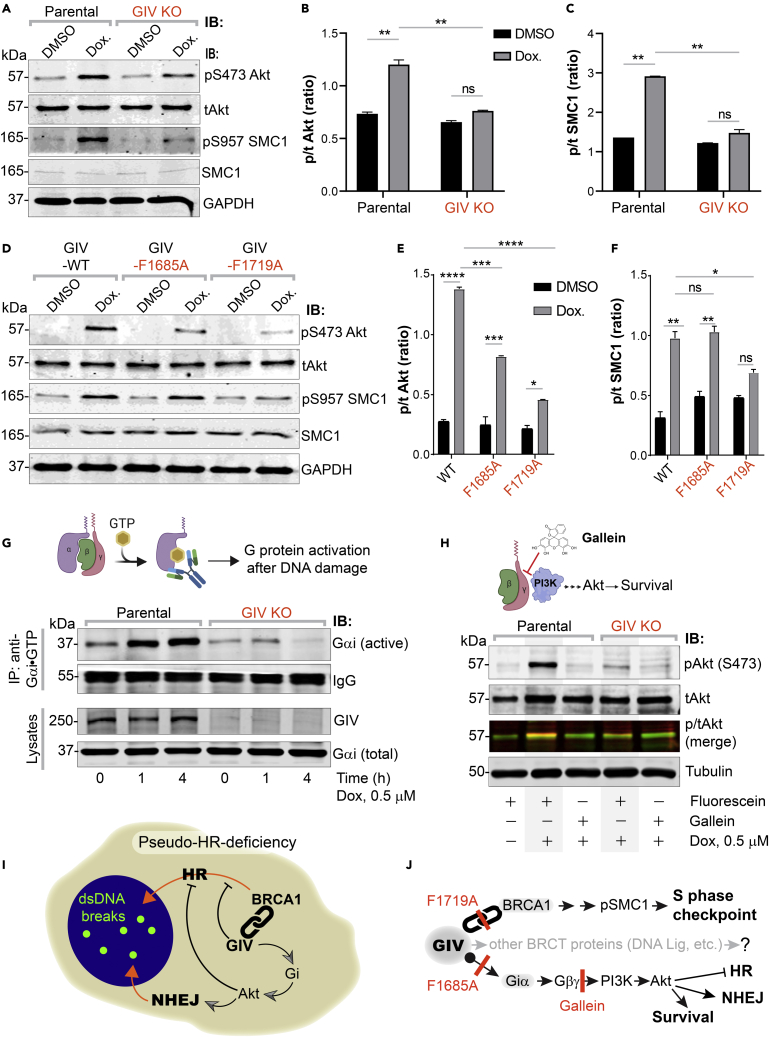
Upon sensing DNA double-strand breaks (DSBs), eukaryotic cells either die or repair DSBs via one of the two competing pathways, i.e., non-homologous end-joining (NHEJ) or homologous recombination (HR). We show that cell fate after DSBs hinges on GIV/Girdin, a guanine nucleotide-exchange modulator of heterotrimeric Giα•βγ protein. GIV suppresses HR by binding and sequestering BRCA1, a key coordinator of multiple steps within the HR pathway, away from DSBs; it does so using a C-terminal motif that binds BRCA1's BRCT-modules via both phospho-dependent and -independent mechanisms. Using another non-overlapping C-terminal motif GIV binds and activates Gi and enhances the "free" Gβγ→PI-3-kinase→Akt pathway, which promotes survival and is known to suppress HR, favor NHEJ. Absence of GIV, or loss of either of its C-terminal motifs enhanced cell death upon genotoxic stress. Because GIV selectively binds other BRCT-containing proteins suggests that G-proteins may fine-tune sensing, repair, and survival after diverse types of DNA damage.
Keywords: Biological sciences; Cell biology; Molecular biology.
© 2023 The Author(s).
Conflict of interest statement
Authors declare no competing interests.
Figures
References
-
- Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017;66:801–817. - PubMed
Grants and funding
- R50 CA221807/CA/NCI NIH HHS/United States
- T32 CA121938/CA/NCI NIH HHS/United States
- R01 CA238042/CA/NCI NIH HHS/United States
- R56 NS073976/NS/NINDS NIH HHS/United States
- R01 CA100768/CA/NCI NIH HHS/United States
- R01 CA160911/CA/NCI NIH HHS/United States
- R01 AI118985/AI/NIAID NIH HHS/United States
- R01 AI141630/AI/NIAID NIH HHS/United States
- R33 CA225549/CA/NCI NIH HHS/United States
- T32 CA067754/CA/NCI NIH HHS/United States
- R01 NS073976/NS/NINDS NIH HHS/United States
- R01 CA238023/CA/NCI NIH HHS/United States
- R01 HL145477/HL/NHLBI NIH HHS/United States
- R01 DK107585/DK/NIDDK NIH HHS/United States
- U01 CA210152/CA/NCI NIH HHS/United States
- UG3 TR002968/TR/NCATS NIH HHS/United States
- R37 CA222563/CA/NCI NIH HHS/United States
- R01 GM117424/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
