

DNA repair in cardiomyocytes is critical for maintaining cardiac function in mice
- PMID: 36756698
- PMCID: PMC10014058
- DOI: 10.1111/acel.13768
DNA repair in cardiomyocytes is critical for maintaining cardiac function in mice
Abstract
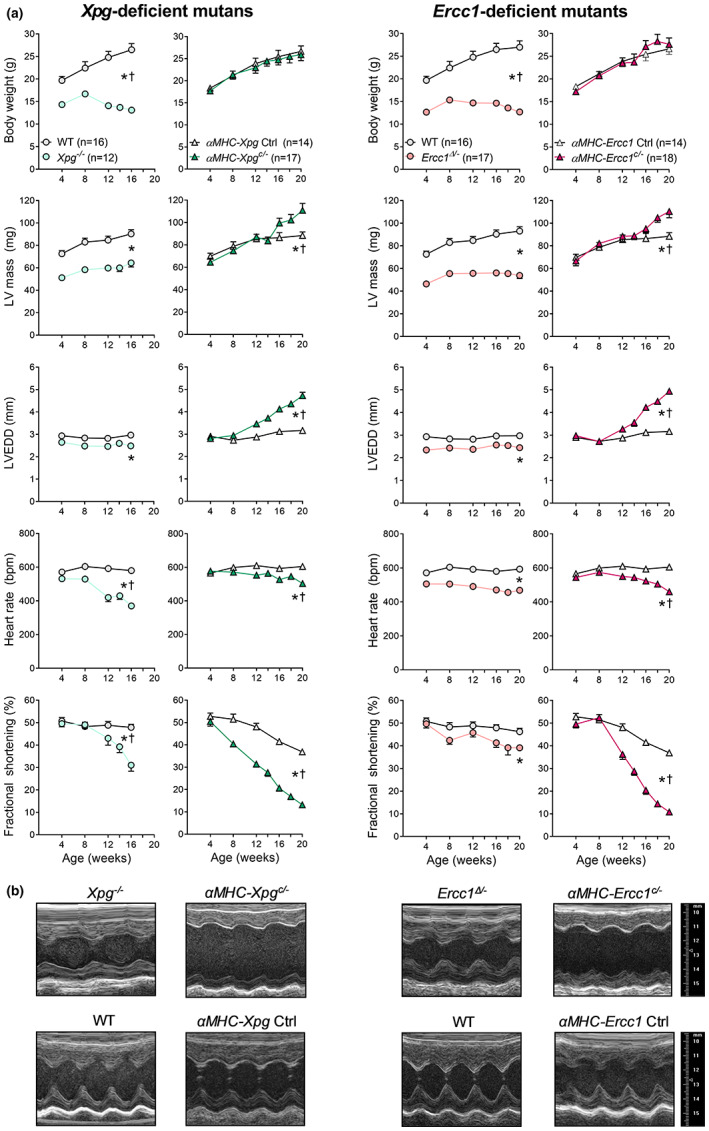
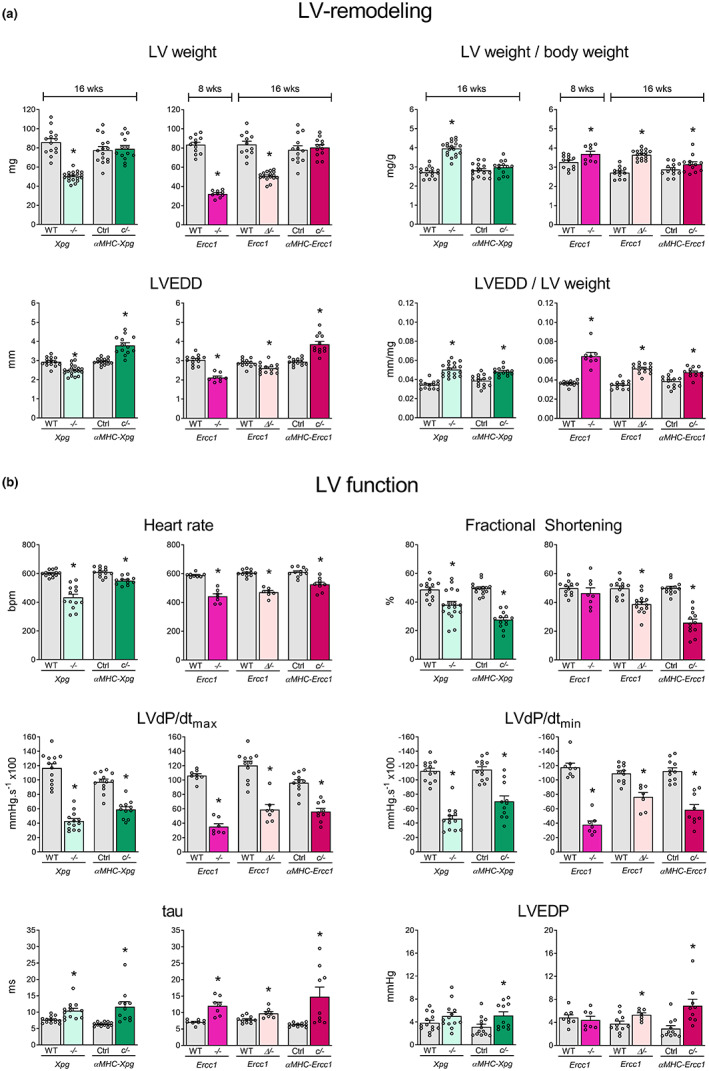
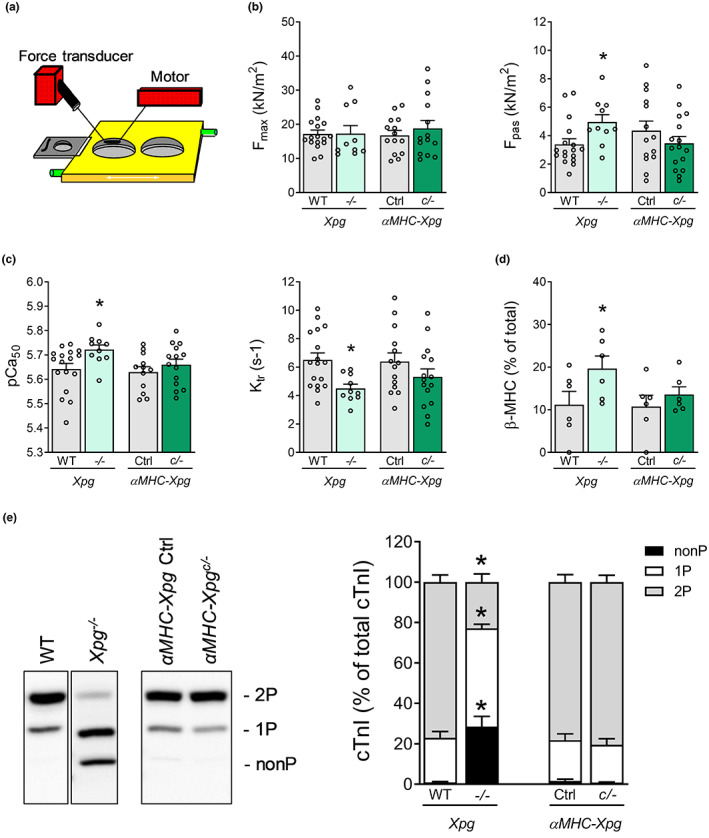
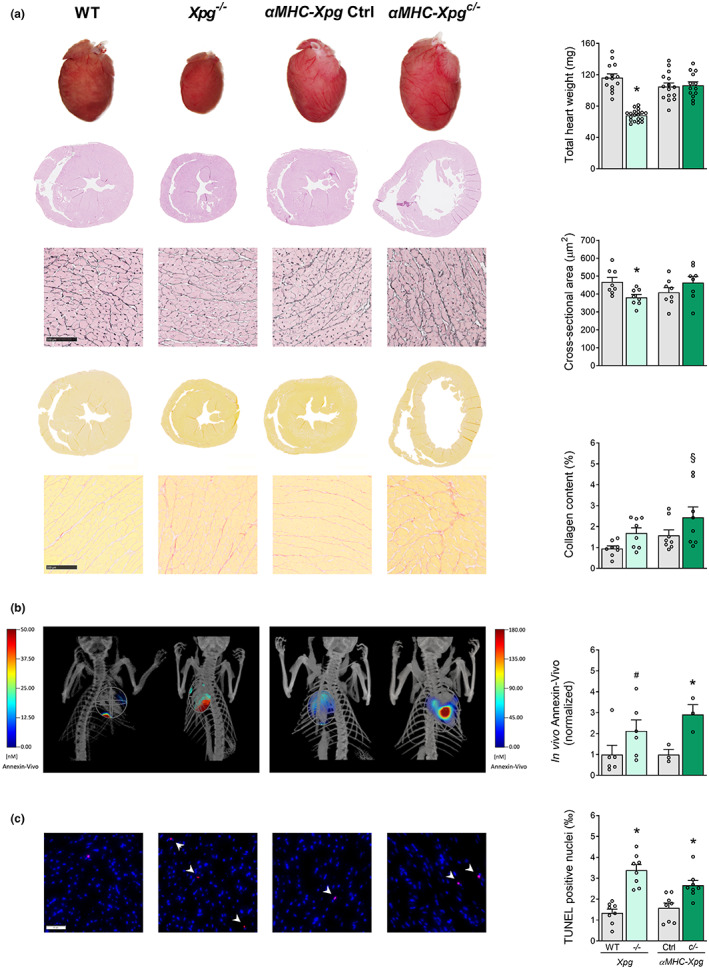
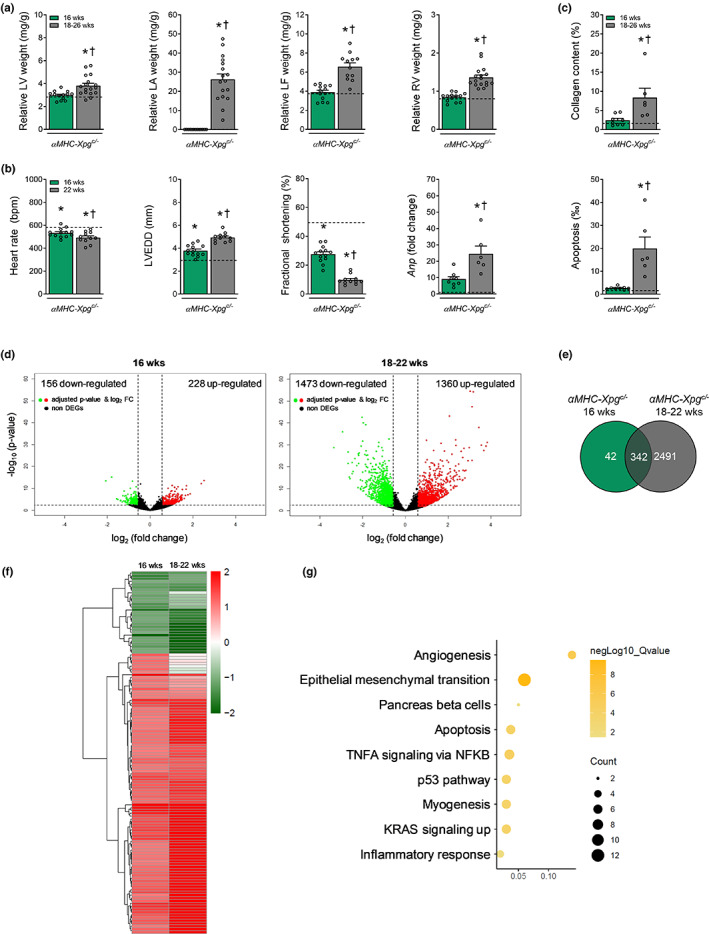
Heart failure has reached epidemic proportions in a progressively ageing population. The molecular mechanisms underlying heart failure remain elusive, but evidence indicates that DNA damage is enhanced in failing hearts. Here, we tested the hypothesis that endogenous DNA repair in cardiomyocytes is critical for maintaining normal cardiac function, so that perturbed repair of spontaneous DNA damage drives early onset of heart failure. To increase the burden of spontaneous DNA damage, we knocked out the DNA repair endonucleases xeroderma pigmentosum complementation group G (XPG) and excision repair cross-complementation group 1 (ERCC1), either systemically or cardiomyocyte-restricted, and studied the effects on cardiac function and structure. Loss of DNA repair permitted normal heart development but subsequently caused progressive deterioration of cardiac function, resulting in overt congestive heart failure and premature death within 6 months. Cardiac biopsies revealed increased oxidative stress associated with increased fibrosis and apoptosis. Moreover, gene set enrichment analysis showed enrichment of pathways associated with impaired DNA repair and apoptosis, and identified TP53 as one of the top active upstream transcription regulators. In support of the observed cardiac phenotype in mutant mice, several genetic variants in the ERCC1 and XPG gene in human GWAS data were found to be associated with cardiac remodelling and dysfunction. In conclusion, unrepaired spontaneous DNA damage in differentiated cardiomyocytes drives early onset of cardiac failure. These observations implicate DNA damage as a potential novel therapeutic target and highlight systemic and cardiomyocyte-restricted DNA repair-deficient mouse mutants as bona fide models of heart failure.
Keywords: DNA damage; DNA repair; apoptosis; cardiac function; congestive heart failure.
© 2023 The Authors. Aging Cell published by Anatomical Society and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Agah, R. , Frenkel, P. A. , French, B. A. , Michael, L. H. , Overbeek, P. A. , & Schneider, M. D. (1997). Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac‐restricted, site‐specific rearrangement in adult ventricular muscle in vivo. The Journal of Clinical Investigation, 100(1), 169–179. 10.1172/JCI119509 - DOI - PMC - PubMed
-
- Ataei Ataabadi, E. , Golshiri, K. , van der Linden, J. , de Boer, M. , Duncker, D. J. , Jüttner, A. , de Vries, R. , van Veghel, R. , van der Pluijm, I. , Dutheil, S. , Chalgeri, S. , Zhang, L. , Lin, A. , Davis, R. E. , Snyder, G. L. , Danser, A. H. J. , & Roks, A. J. M. (2021). Vascular ageing features caused by selective DNA damage in smooth muscle cell. Oxidative Medicine and Cellular Longevity, 2021, 2308317. 10.1155/2021/2308317 - DOI - PMC - PubMed
-
- Barnhoorn, S. , Uittenboogaard, L. M. , Jaarsma, D. , Vermeij, W. P. , Tresini, M. , Weymaere, M. , Menoni, H. , Brandt, R. M. , de Waard, M. C. , Botter, S. M. , Sarker, A. H. , Jaspers, N. G. , van der Horst, G. , Cooper, P. K. , Hoeijmakers, J. H. , & van der Pluijm, I. (2014). Cell‐autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genetics, 10(10), e1004686. 10.1371/journal.pgen.1004686 - DOI - PMC - PubMed
-
- Bartunek, J. , Vanderheyden, M. , Knaapen, M. W. , Tack, W. , Kockx, M. M. , & Goethals, M. (2002). Deoxyribonucleic acid damage/repair proteins are elevated in the failing human myocardium due to idiopathic dilated cardiomyopathy. Journal of the American College of Cardiology, 40(6), 1097–1103; discussion 1104‐1095. 10.1016/s0735-1097(02)02122-8 - DOI - PubMed
-
- Bautista‐Nino, P. K. , Portilla‐Fernandez, E. , Rubio‐Beltran, E. , van der Linden, J. J. , de Vries, R. , van Veghel, R. , de Boer, M. , Durik, M. , Ridwan, Y. , Brandt, R. , Essers, J. , Menzies, R. I. , Thomas, R. , de Bruin, A. , Duncker, D. J. , van HMM, B. , Ghanbari, M. , JHJ, H. , Sedlacek, R. , … Roks, A. J. M. (2020). Local endothelial DNA repair deficiency causes aging‐resembling endothelial‐specific dysfunction. Clinical Science, 134(7), 727–746. 10.1042/CS20190124 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
