

Multiplexed Transgenic Selection and Counterselection Strategies to Expedite Genetic Manipulation Workflows Using Drosophila melanogaster
- PMID: 36757287
- PMCID: PMC9923875
- DOI: 10.1002/cpz1.652
Multiplexed Transgenic Selection and Counterselection Strategies to Expedite Genetic Manipulation Workflows Using Drosophila melanogaster
Abstract
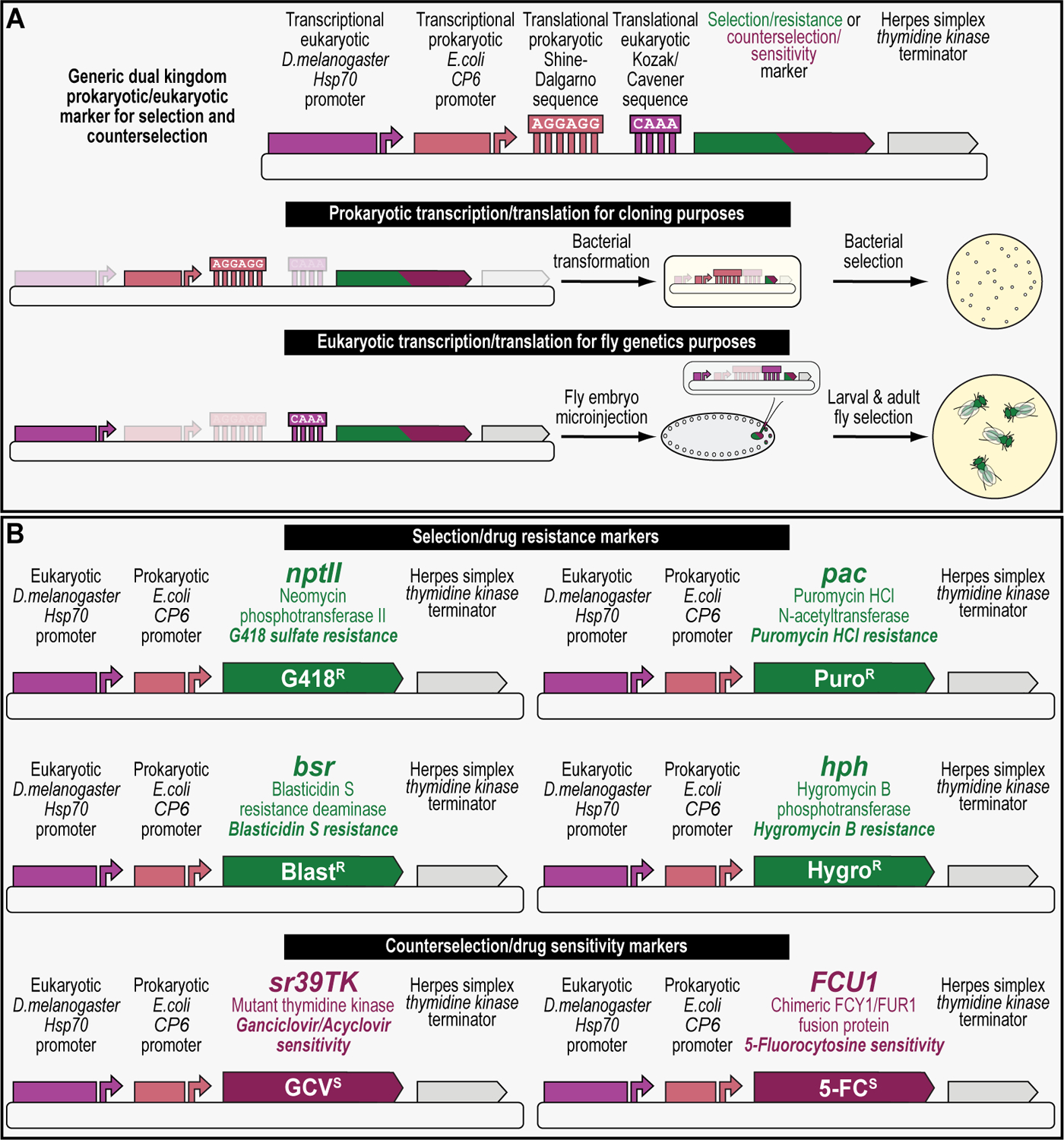
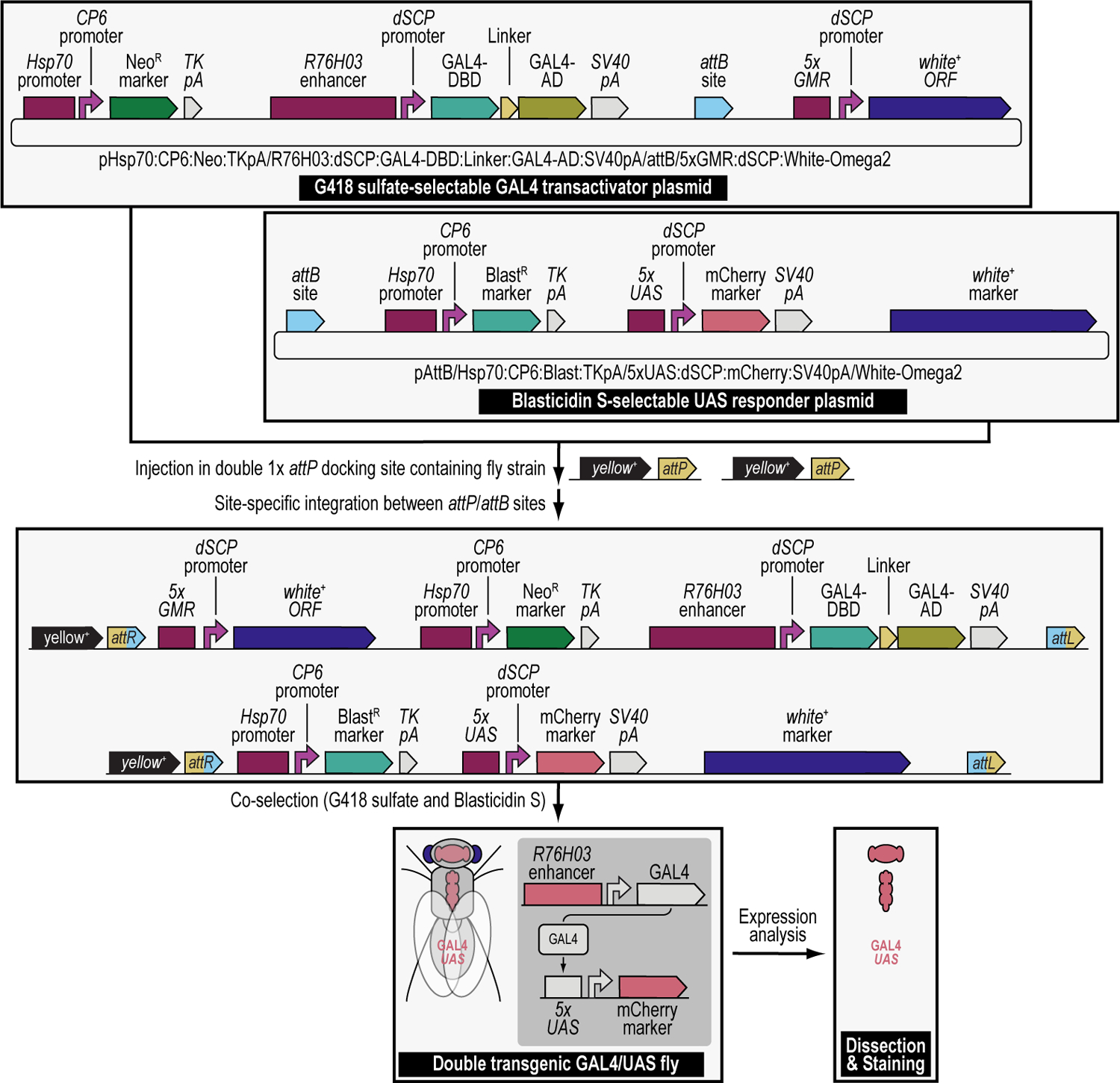
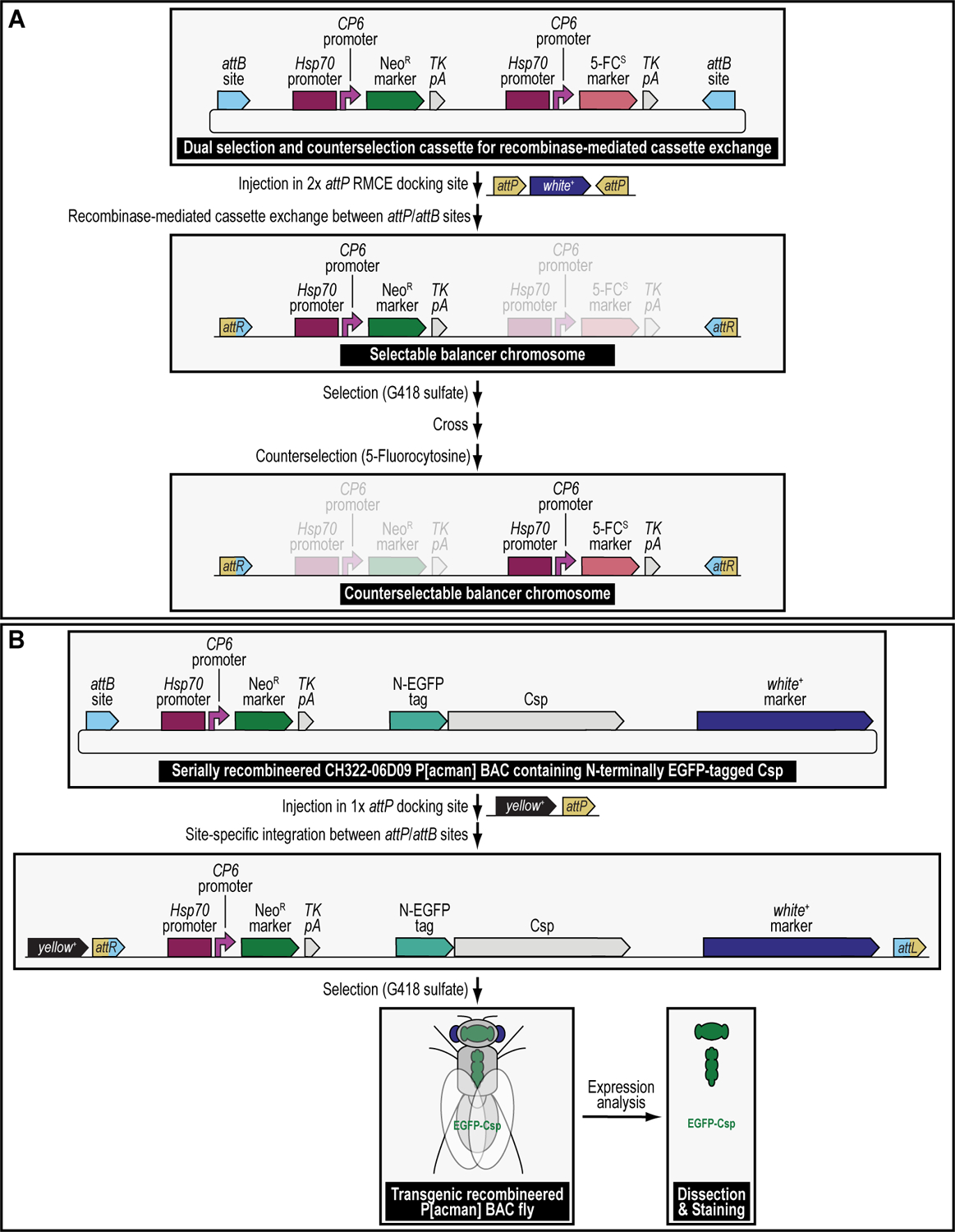
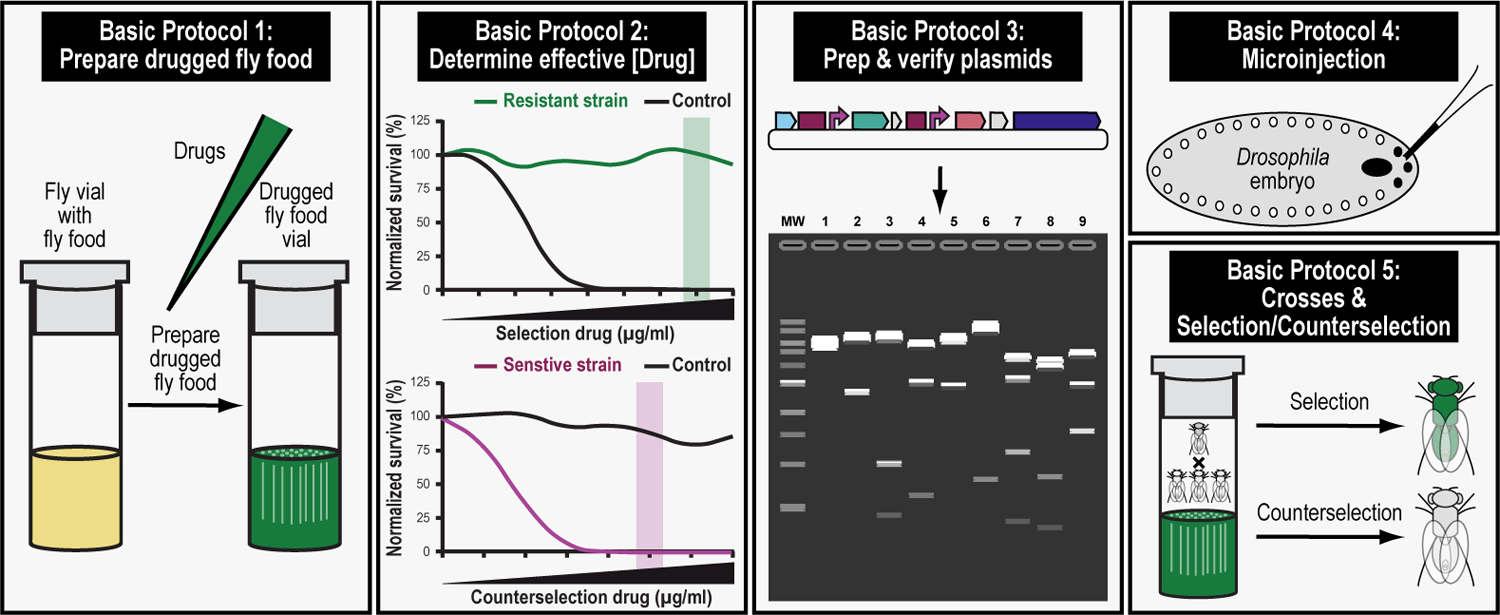
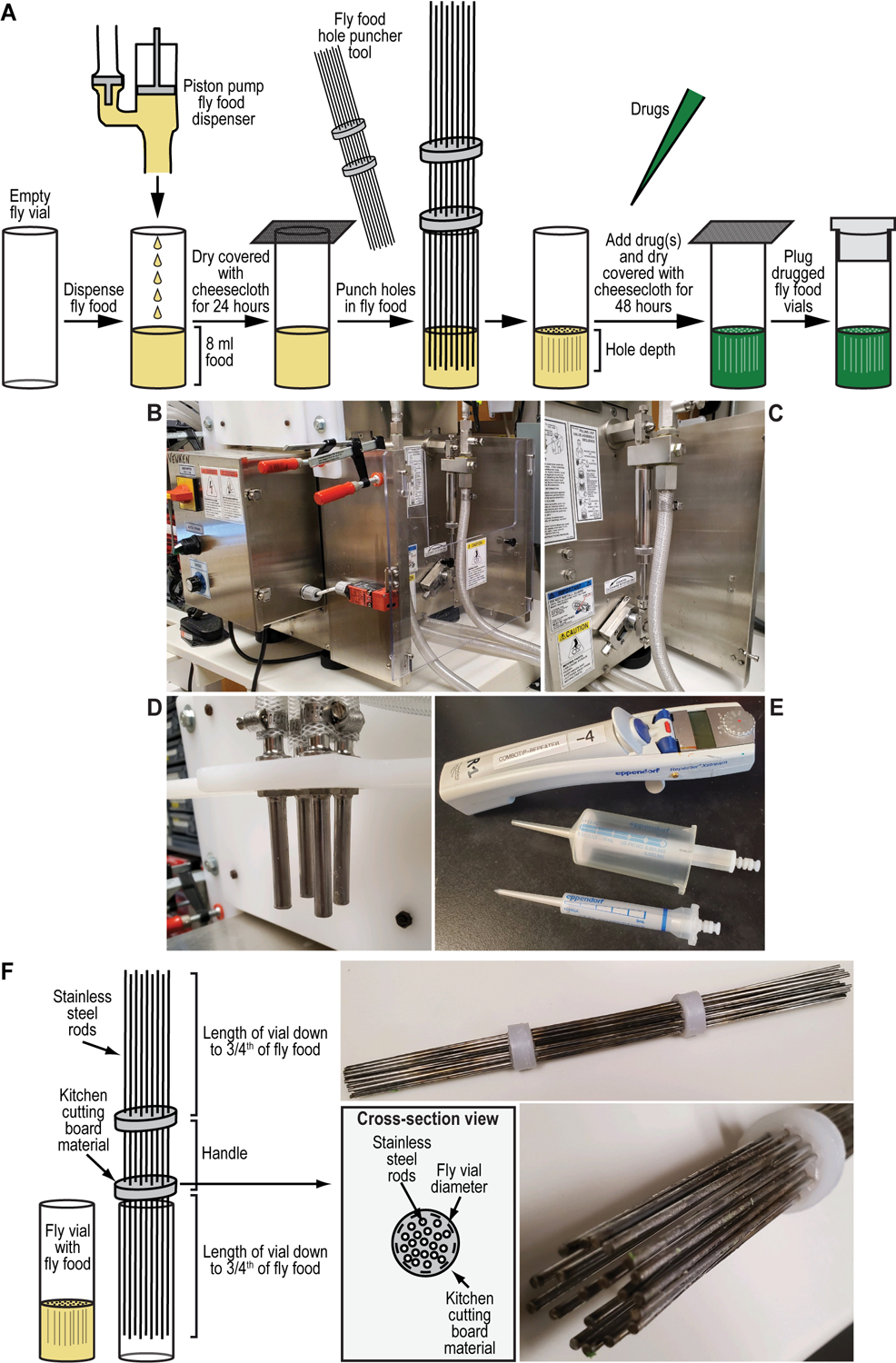
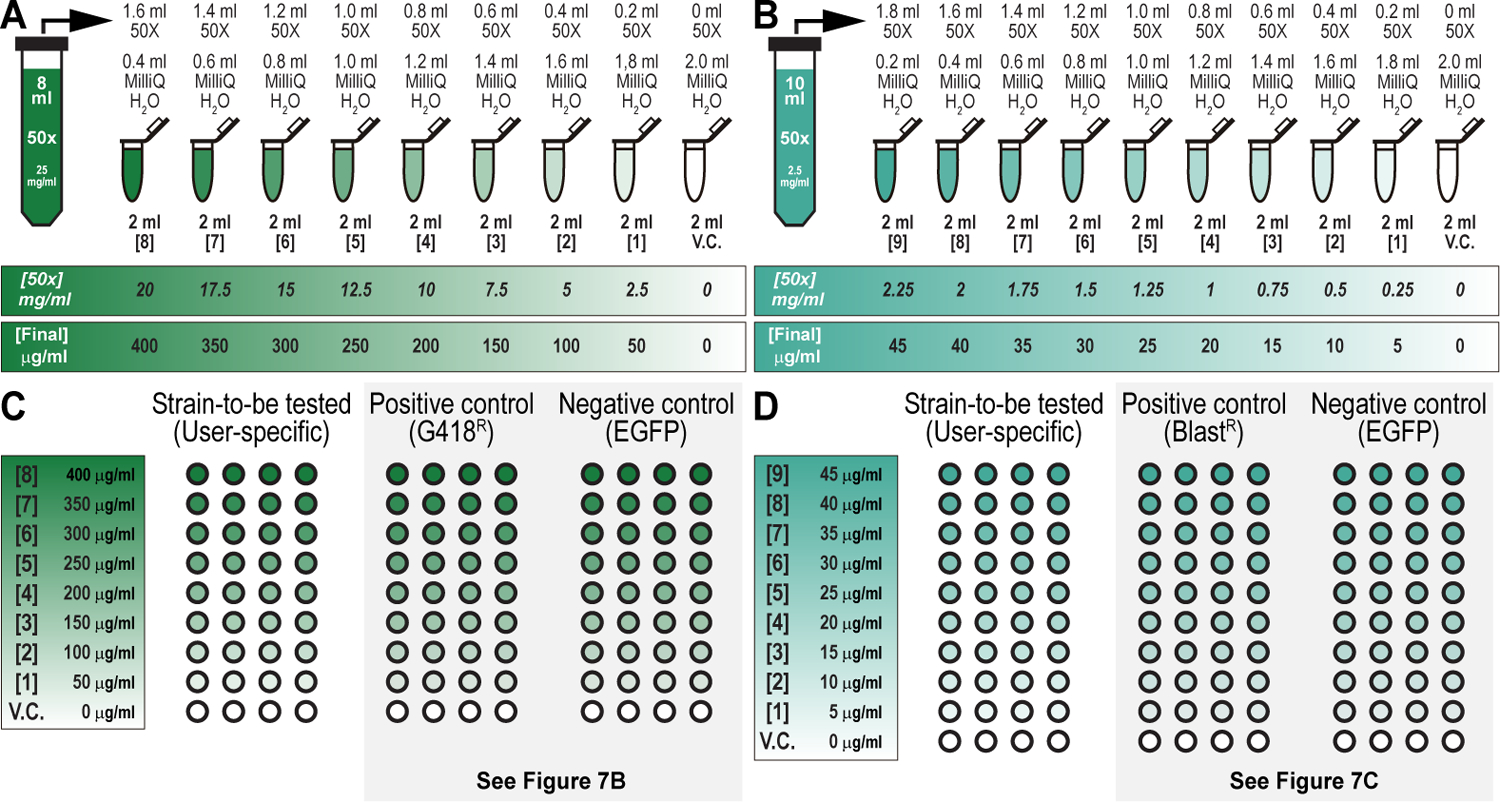
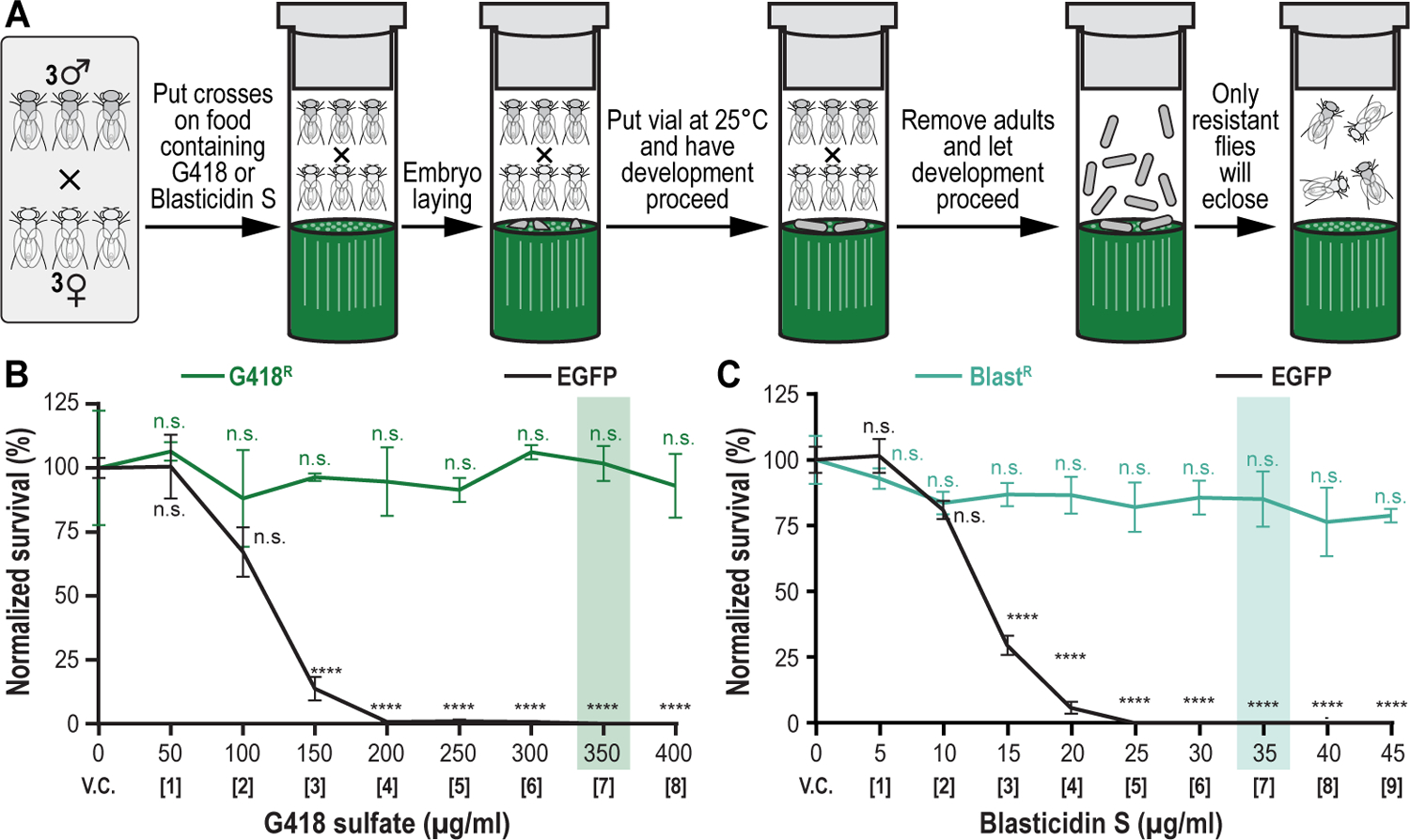
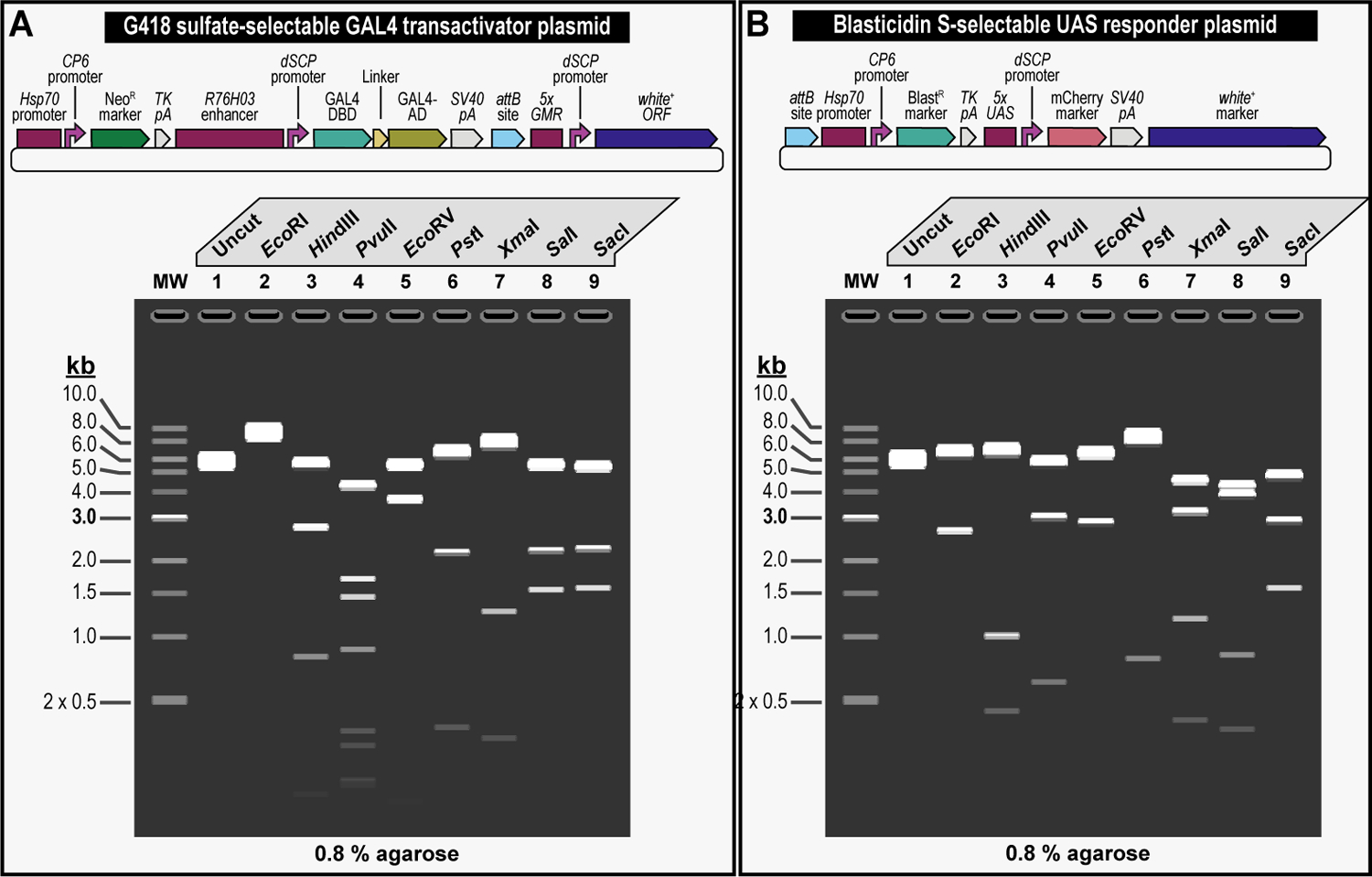
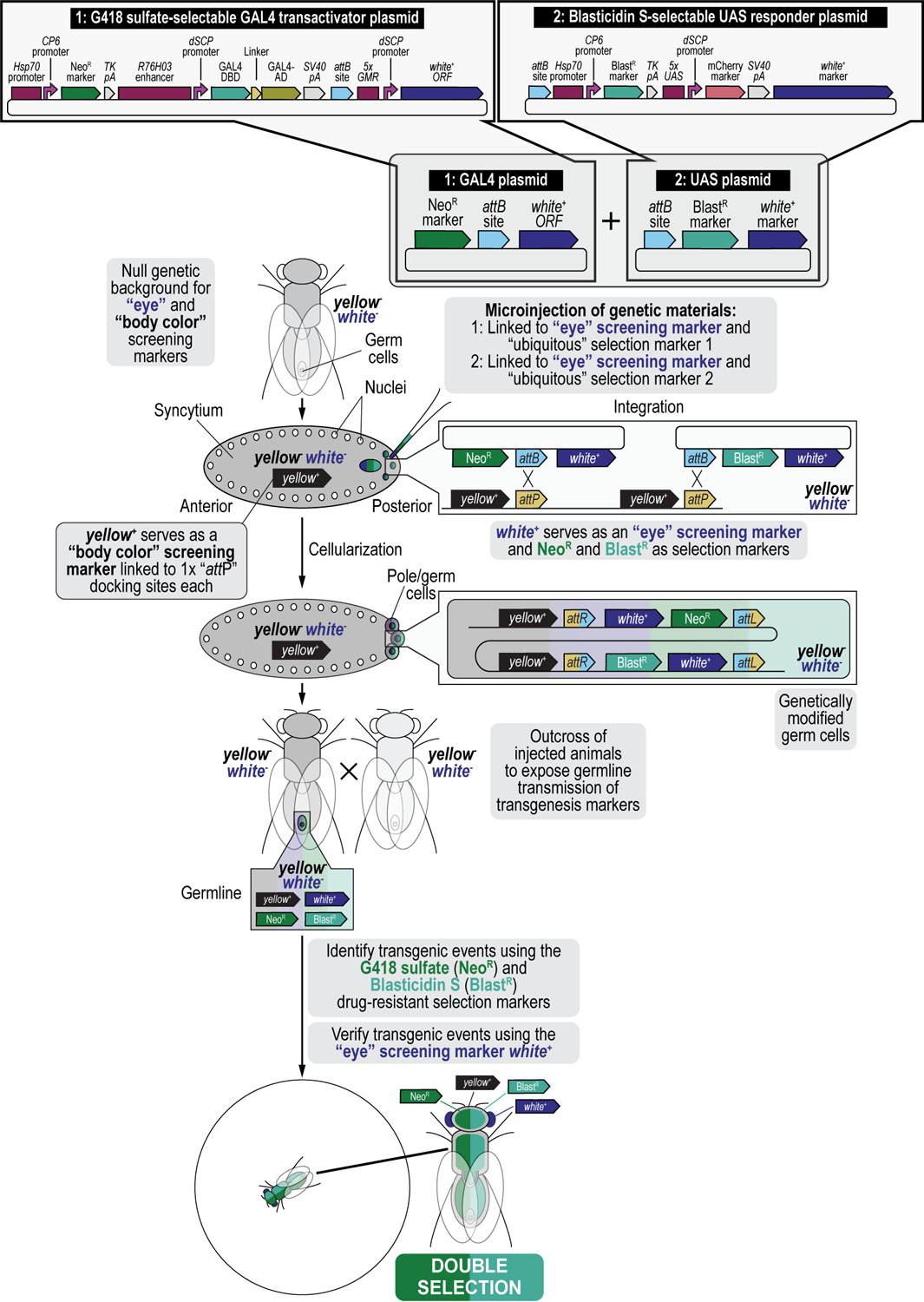
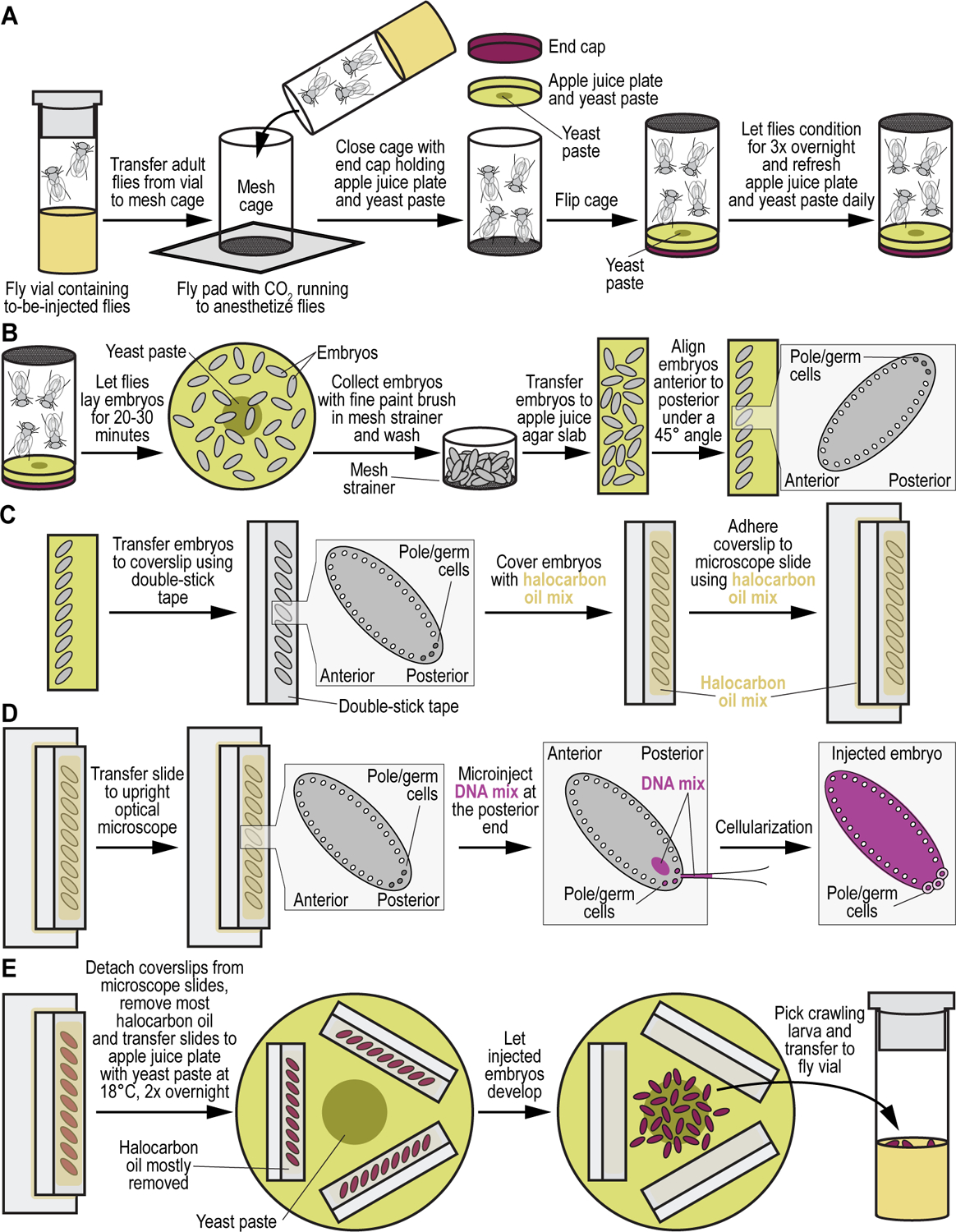
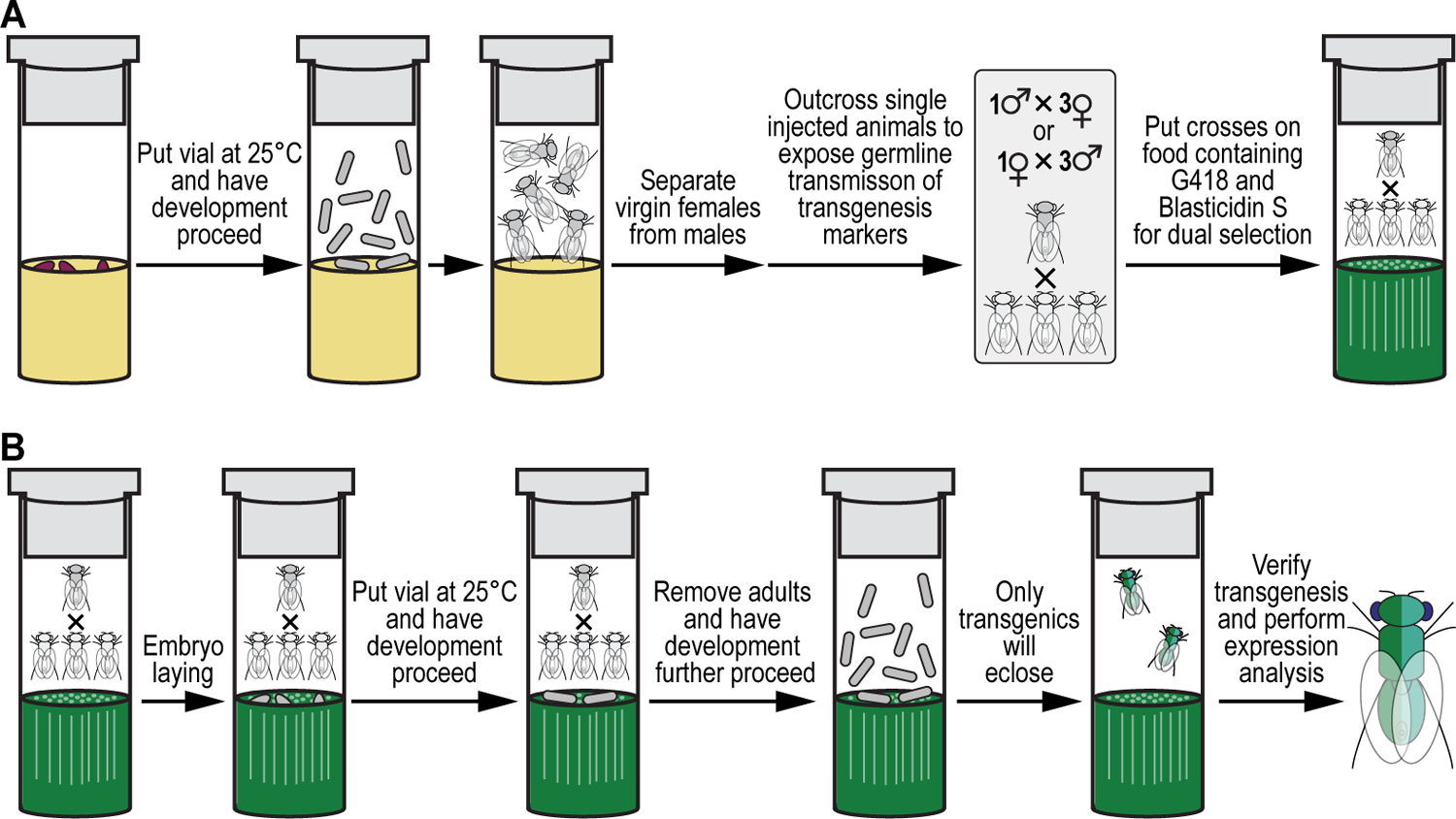
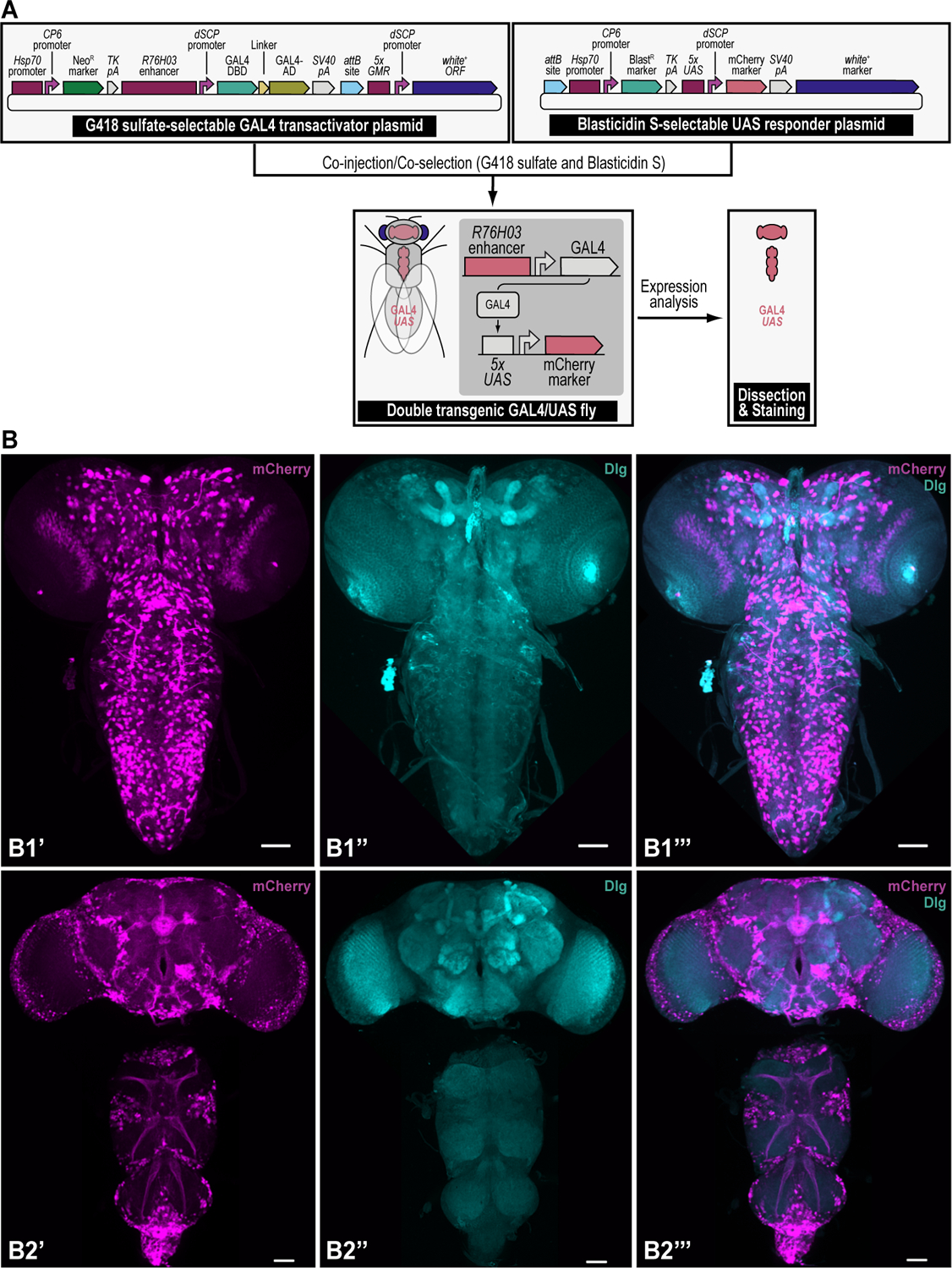
We recently described a set of four selectable and two counterselectable markers that provide resistance and sensitivity, respectively, against their corresponding drugs using the model organism Drosophila melanogaster. The four selectable markers provide animals with resistance against G418 sulfate, puromycin HCl, blasticidin S, or hygromycin B, whereas the two counterselection markers make animals sensitive to ganciclovir/acyclovir or 5-fluorocytosine. Unlike classical phenotypic markers, whether visual or fluorescent, which require extensive screening of progeny of a genetic cross for desired genotypes, resistance and sensitivity markers eliminate this laborious procedure by directly selecting for, or counterselecting against, the desired genotypes. We demonstrated the usefulness of these markers with three applications: 1) generating dual transgenic animals for binary overexpression (e.g., GAL4/UAS) analysis in a single step through the process of co-injection, followed by co-selection resulting in co-transgenesis; 2) obtaining balancer chromosomes that are both selectable and counterselectable to manipulate crossing schemes for, or against, the presence of the modified balancer chromosome; and 3) making both selectable and fluorescently tagged P[acman] BAC transgenic animals for gene expression and proteomic analysis. Here, we describe detailed procedures for how to use these drug-based selection and counterselection markers in the fruit fly D. melanogaster when making dual transgenic animals for binary overexpression as an example. Dual transgenesis integrates site-specifically into two sites in the genome in a single step, namely both components of the binary GAL4/UAS overexpression system, via a G418 sulfate-selectable GAL4 transactivator plasmid and a blasticidin S-selectable UAS responder plasmid. The process involves co-injecting the two plasmids, followed by co-selection using G418 sulfate and blasticidin S, resulting in co-transgenesis of the two plasmids in the fly genome. We demonstrate the functionality of the procedure based on the expression pattern obtained after dual transgenesis of the two plasmids. We provide protocols on how to prepare drugged fly food vials, determine the effective drug concentration for markers used during transgenic selection and counterselection strategies, and prepare and confirm plasmid DNA for microinjection, followed by the microinjection procedure itself and setting up crossing schemes to isolate desired progeny through selection and/or counterselection. These protocols can be easily adapted to any combination of the six selectable and counterselectable markers we described or any new marker that is resistant or sensitive to a novel drug. Protocols on how to build plasmids by synthetic-assembly DNA cloning or modify plasmids by serial recombineering to perform a plethora of selection, counterselection, or any other genetic strategies are presented in two accompanying Current Protocols articles. © 2023 Wiley Periodicals LLC. Basic Protocol 1: Preparing drugged fly food vials for transgenic selection and counterselection strategies using D. melanogaster Basic Protocol 2: Determining the effective drug concentration for resistance and sensitivity markers used during transgenic selection and counterselection strategies using D. melanogaster Basic Protocol 3: Preparing and confirming plasmid DNA for microinjection to perform transgenic selection and counterselection strategies using D. melanogaster Basic Protocol 4: Microinjecting plasmid DNA into fly embryos to perform transgenic selection and counterselection strategies using D. melanogaster Basic Protocol 5: Crossing schemes to isolate desired progeny through transgenic selection and counterselection strategies using D. melanogaster.
Keywords: Drosophila melanogaster; counterselection; genetic manipulation; genetics; multiplexed; selection; transgenesis.
© 2023 Wiley Periodicals LLC.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest
Figures
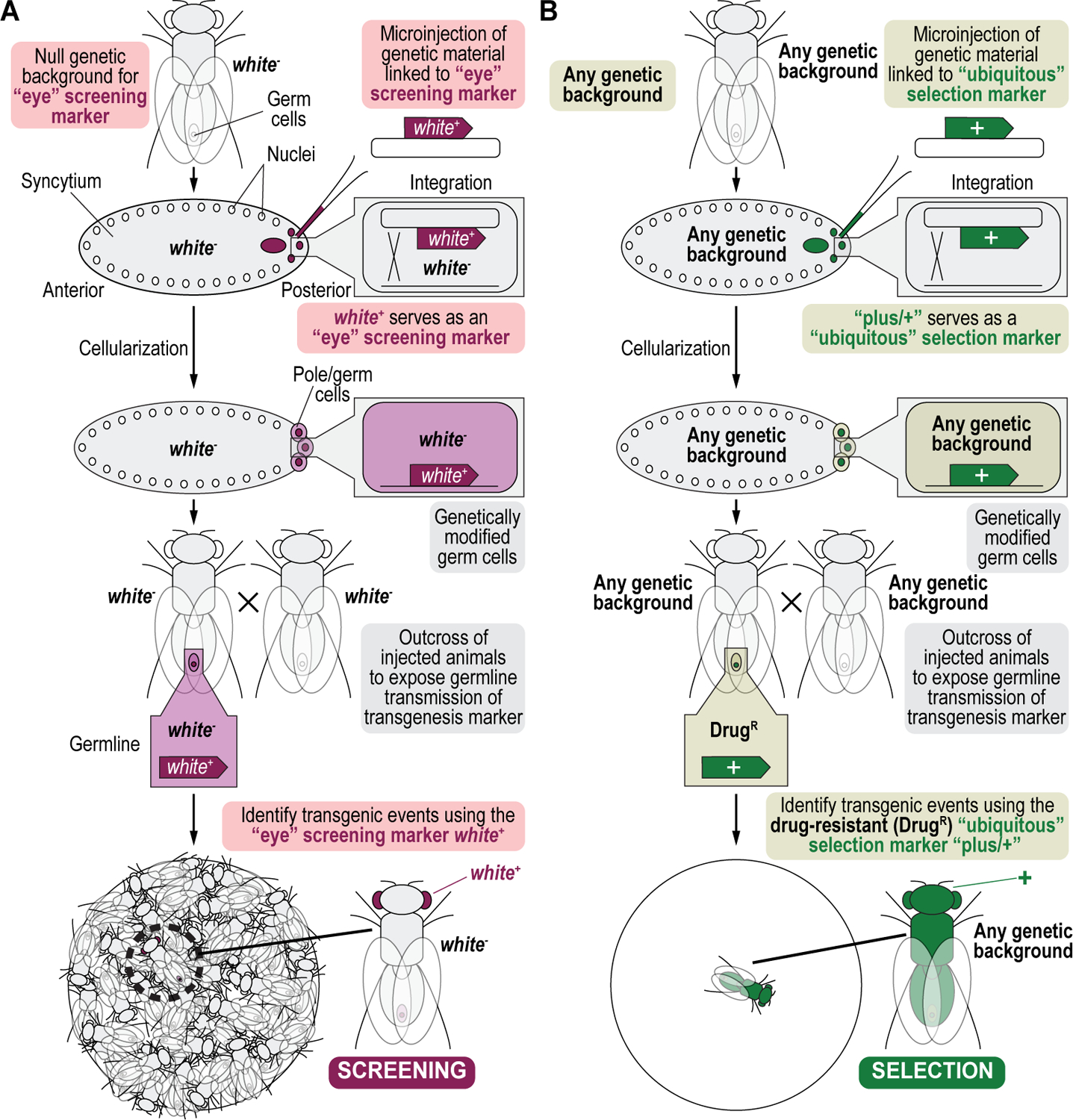
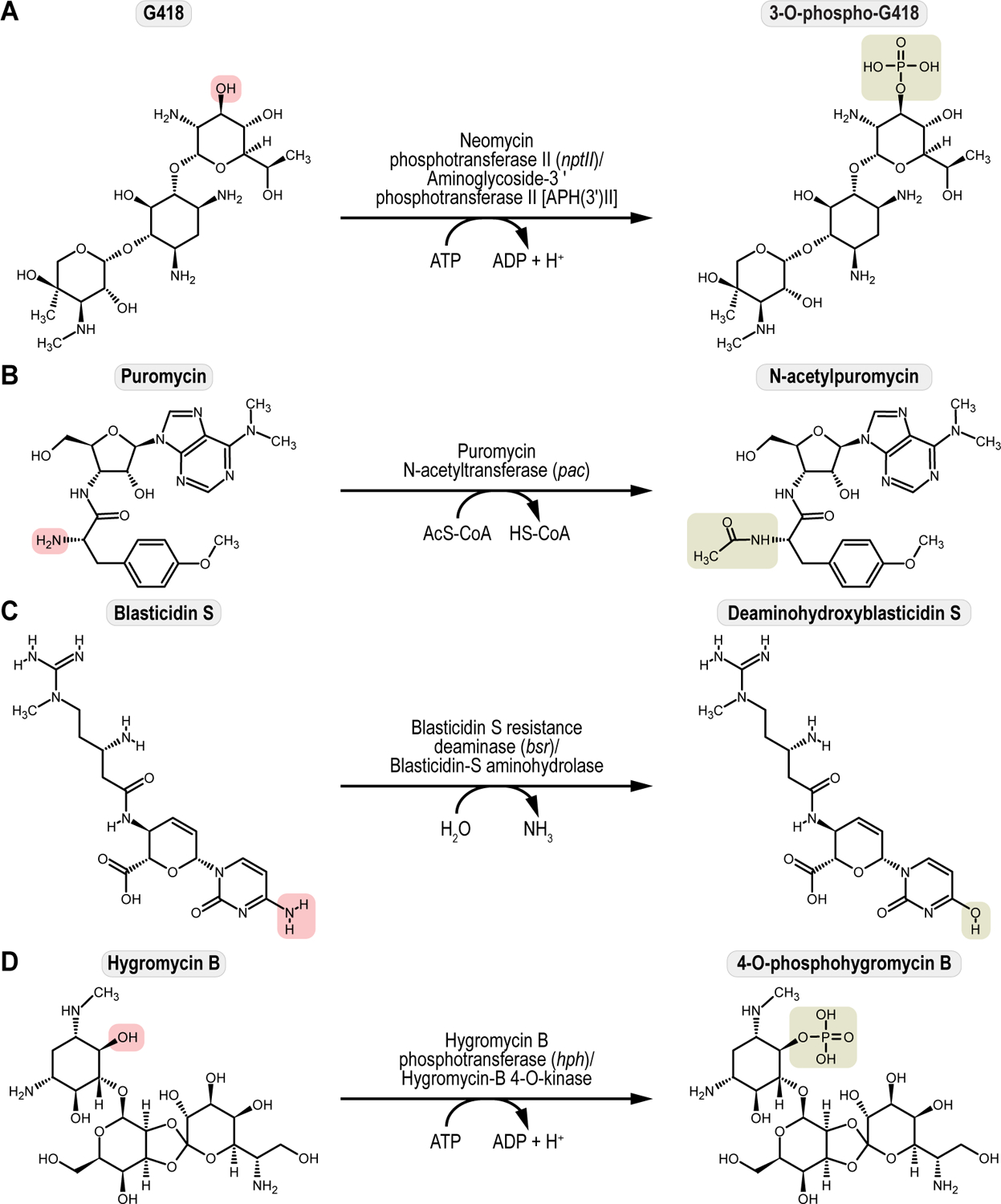
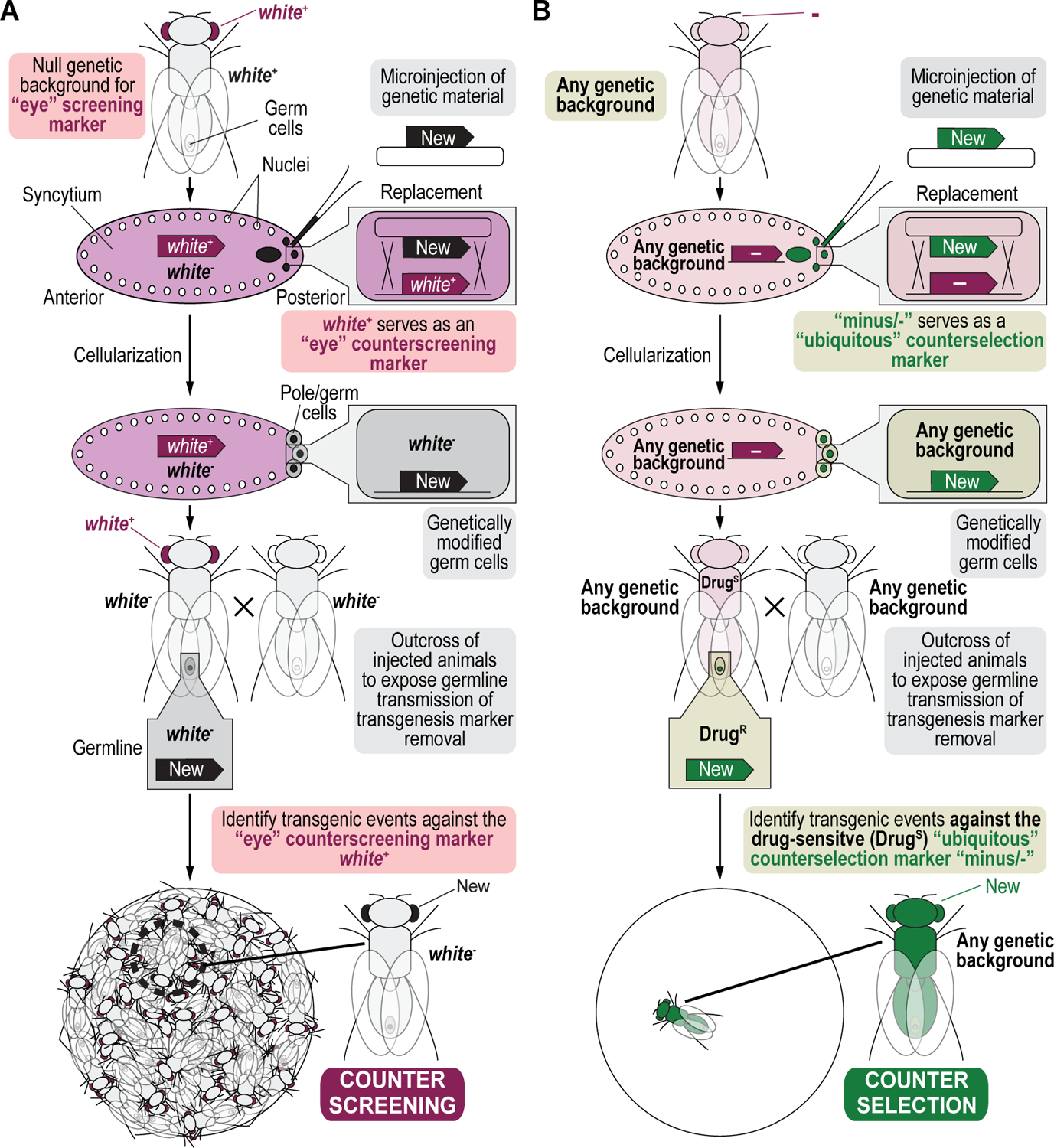
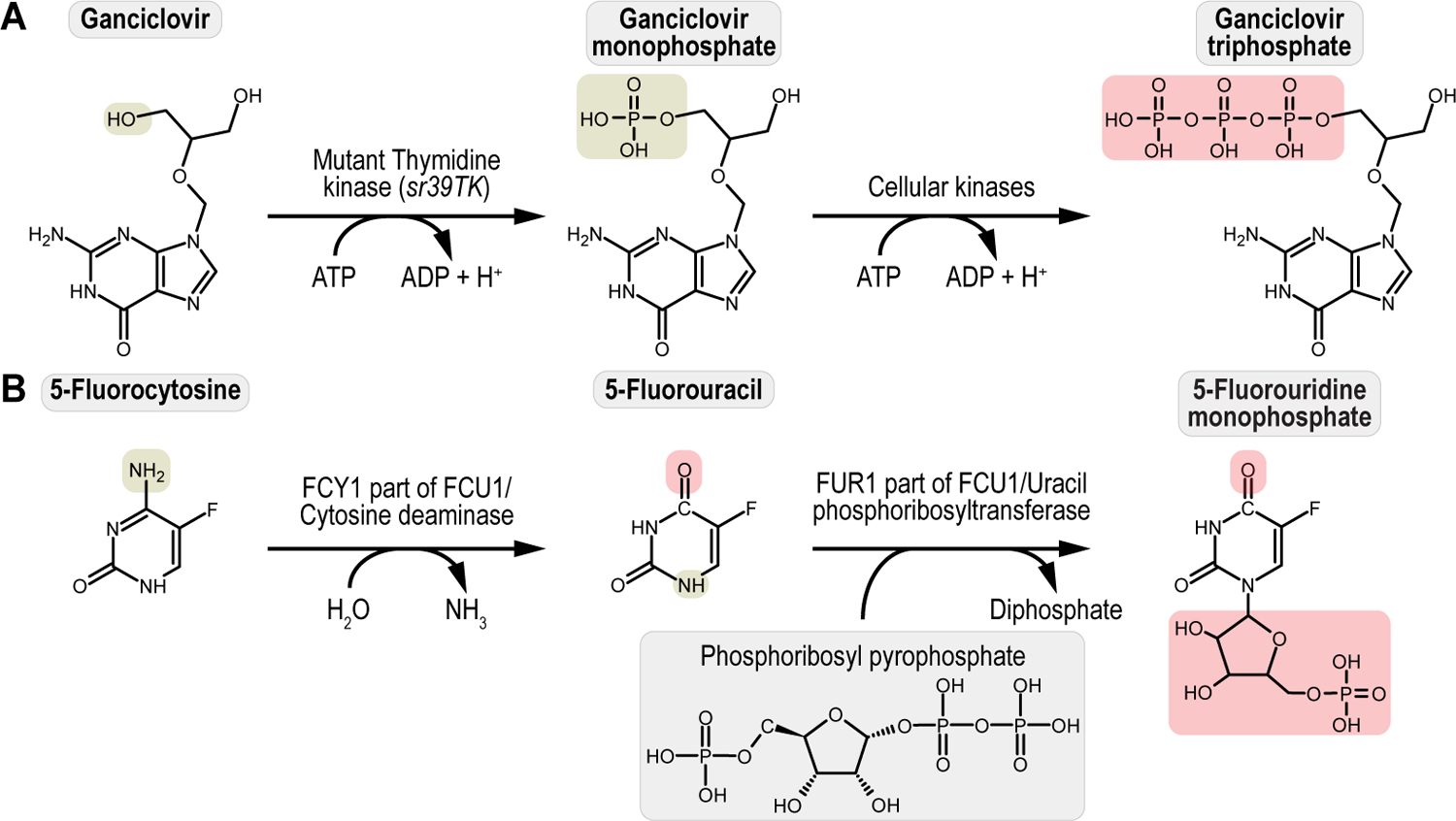
References
LITERATURE CITED
-
- Benedict MQ, Salazar CE, and Collins FH 1995. A new dominant selectable marker for genetic transformation; Hsp70-opd. Insect Biochemistry and Molecular Biology 25:1061–1065. - PubMed
-
- Bier E 2005. Drosophila, the golden bug, emerges as a tool for human genetics. Nature Reviews Genetics 6:9–23. - PubMed
INTERNET RESOURCES
-
- Public plasmid repository, Addgene. https://www.addgene.org/
-
- DNA manipulation software, SnapGene. https://www.snapgene.com/
-
- Public Drosophila fly strain repository, Bloomington Drosophila Stock Center. https://bdsc.indiana.edu/
-
- Free chemical structure database, ChemSpider. https://www.chemspider.com/
-
- Chemical structure drawing program with a free academic version, ChemSketch. https://www.acdlabs.com/
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
