

Multi-'omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients
- PMID: 36758521
- PMCID: PMC10353054
- DOI: 10.1016/j.chom.2023.01.001
Multi-'omics of gut microbiome-host interactions in short- and long-term myalgic encephalomyelitis/chronic fatigue syndrome patients
Abstract
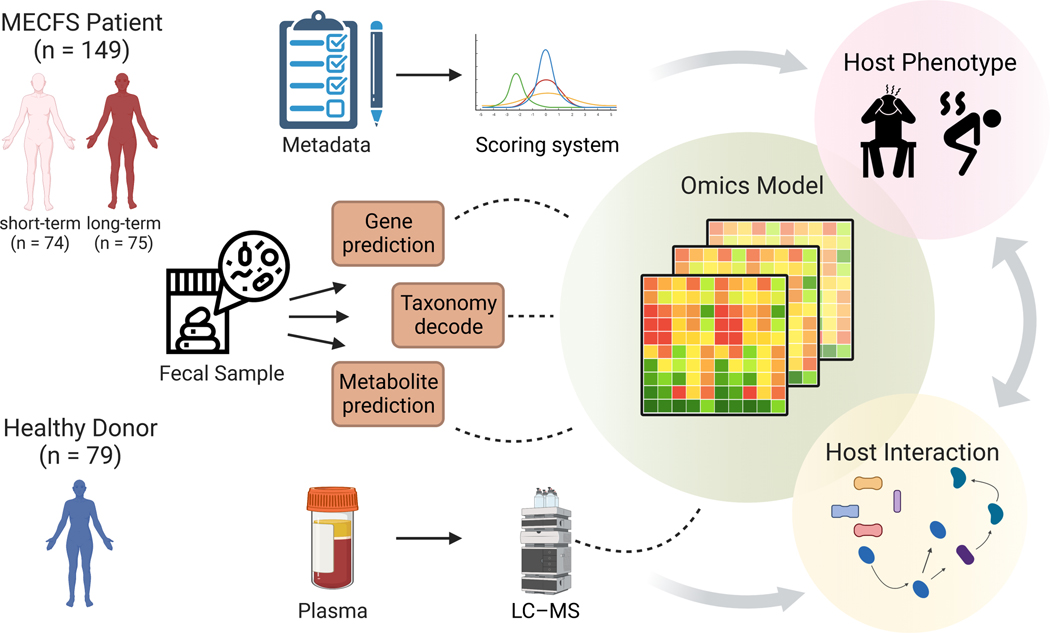
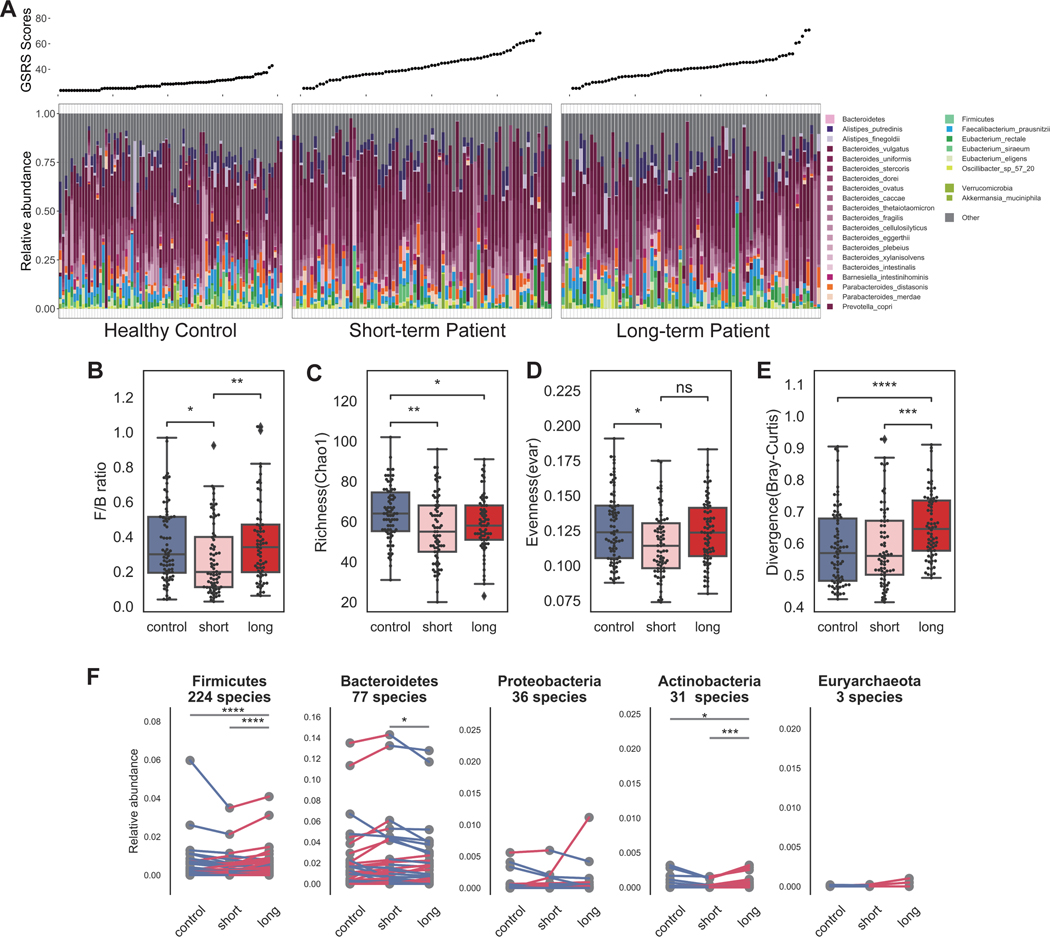
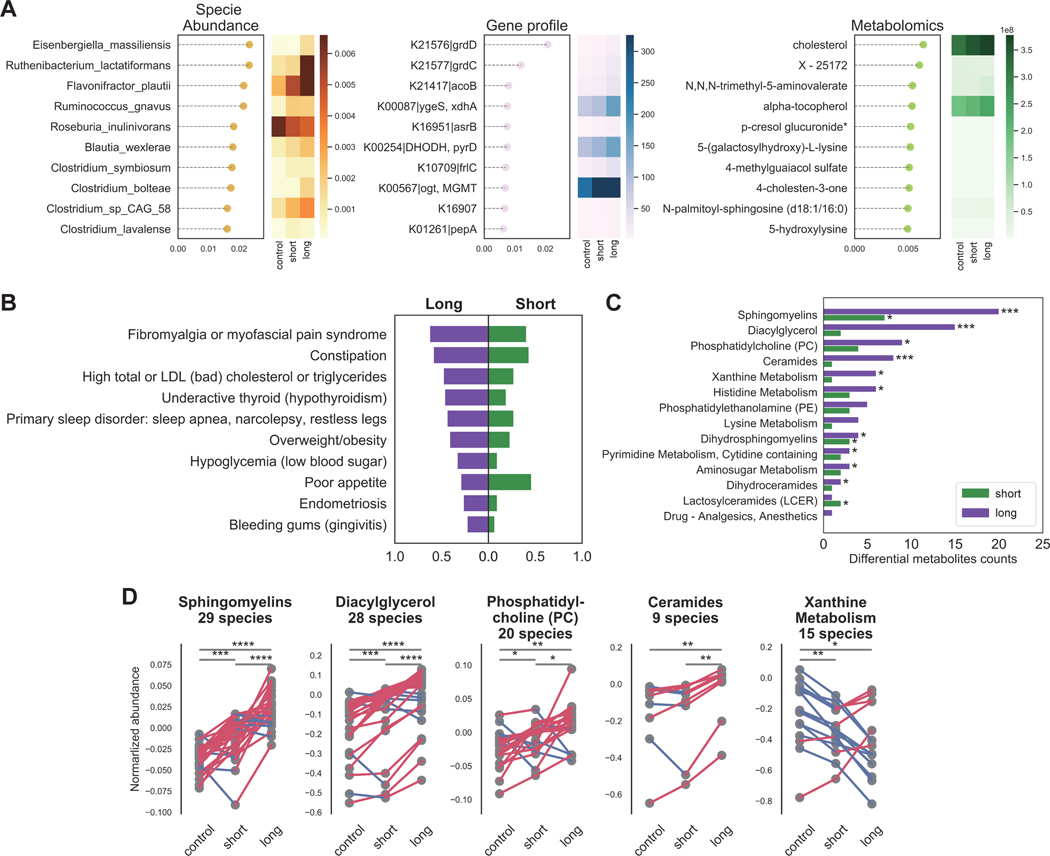
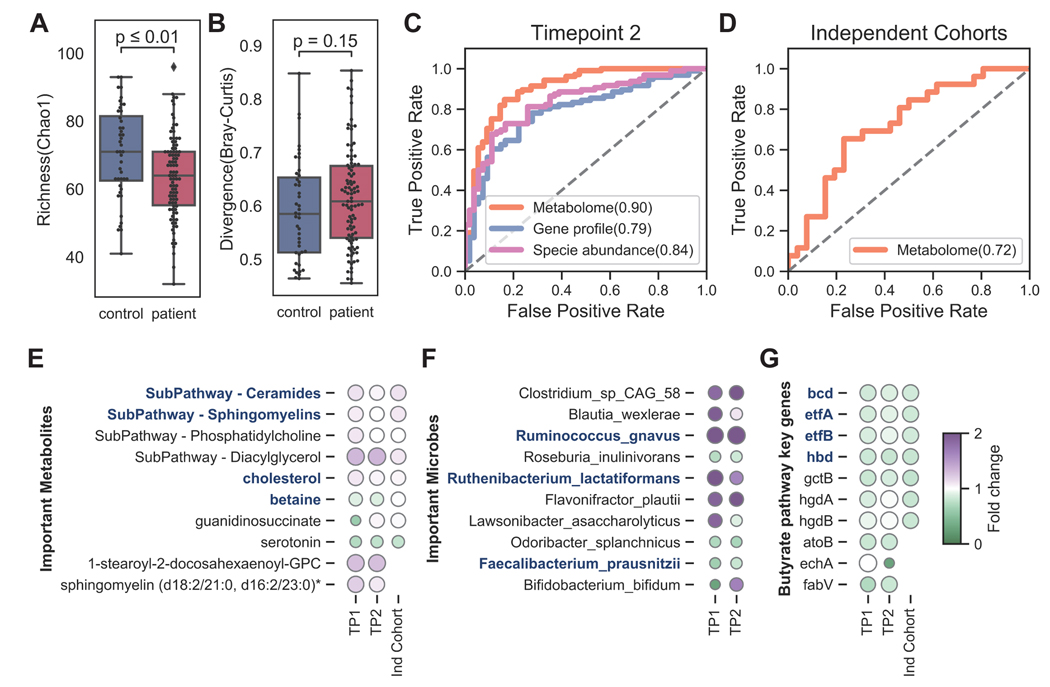
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a complex, debilitating disorder manifesting as severe fatigue and post-exertional malaise. The etiology of ME/CFS remains elusive. Here, we present a deep metagenomic analysis of stool combined with plasma metabolomics and clinical phenotyping of two ME/CFS cohorts with short-term (<4 years, n = 75) or long-term disease (>10 years, n = 79) compared with healthy controls (n = 79). First, we describe microbial and metabolomic dysbiosis in ME/CFS patients. Short-term patients showed significant microbial dysbiosis, while long-term patients had largely resolved microbial dysbiosis but had metabolic and clinical aberrations. Second, we identified phenotypic, microbial, and metabolic biomarkers specific to patient cohorts. These revealed potential functional mechanisms underlying disease onset and duration, including reduced microbial butyrate biosynthesis and a reduction in plasma butyrate, bile acids, and benzoate. In addition to the insights derived, our data represent an important resource to facilitate mechanistic hypotheses of host-microbiome interactions in ME/CFS.
Keywords: ME/CFS; biomarker; gut microbiome; metabolomics; metagenomics; multi-‘omics.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests S.D.V. is affiliated and has a financial interest with The BioCollective, a company that provided the BioCollector, the collection kit used for at home stool collection discussed in this manuscript.
Figures



Comment in
-
Revealing gut microbiome associations with CFS.Cell Host Microbe. 2023 Feb 8;31(2):171-172. doi: 10.1016/j.chom.2023.01.011. Cell Host Microbe. 2023. PMID: 36758517
-
Deficient butyrate-producing capacity in the gut microbiome is associated with bacterial network disturbances and fatigue symptoms in ME/CFS.Cell Host Microbe. 2023 Feb 8;31(2):288-304.e8. doi: 10.1016/j.chom.2023.01.004. Cell Host Microbe. 2023. PMID: 36758522 Free PMC article.
References
-
- Pheby DFH, Araja D, Berkis U, Brenna E, Cullinan J, de Korwin J-D, Gitto L, Hughes DA, Hunter RM, Trepel D, et al. (2021). A Literature Review of GP Knowledge and Understanding of ME/CFS: A Report from the Socioeconomic Working Group of the European Network on ME/CFS (EUROMENE). Medicina (Mex.) 57, 7. 10.3390/medicina57010007. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
