

A reversible SRC-relayed COX2 inflammatory program drives resistance to BRAF and EGFR inhibition in BRAFV600E colorectal tumors
- PMID: 36759733
- PMCID: PMC9970872
- DOI: 10.1038/s43018-022-00508-5
A reversible SRC-relayed COX2 inflammatory program drives resistance to BRAF and EGFR inhibition in BRAFV600E colorectal tumors
Abstract
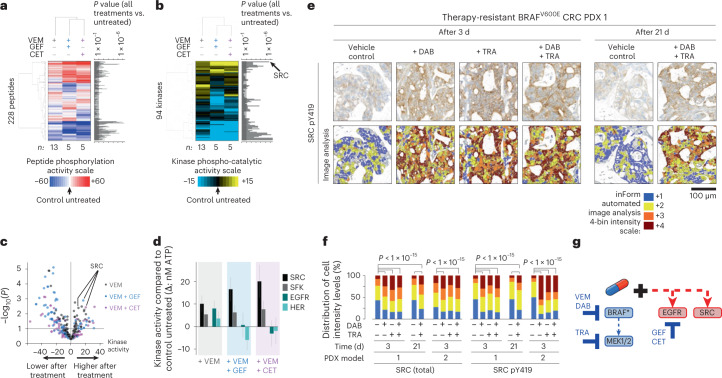
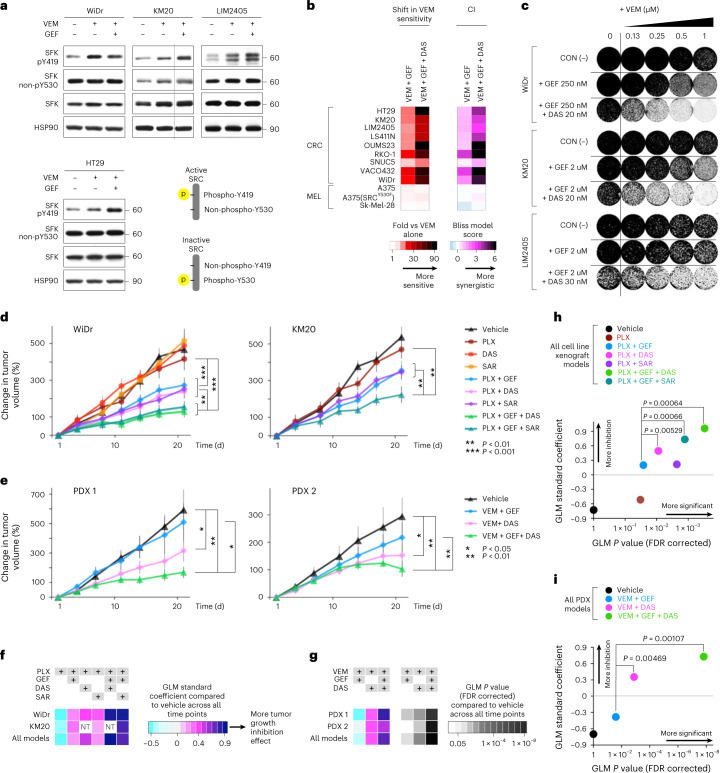
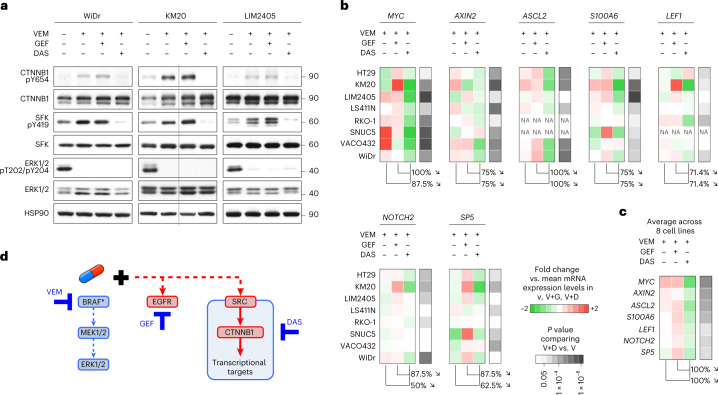
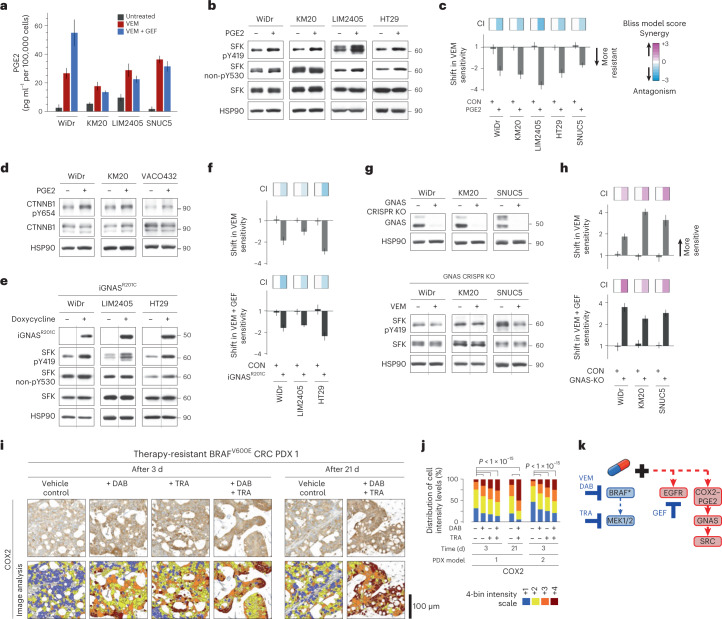
BRAFV600E mutation confers a poor prognosis in metastatic colorectal cancer (CRC) despite combinatorial targeted therapies based on the latest understanding of signaling circuitry. To identify parallel resistance mechanisms induced by BRAF-MEK-EGFR co-targeting, we used a high-throughput kinase activity mapping platform. Here we show that SRC kinases are systematically activated in BRAFV600E CRC following targeted inhibition of BRAF ± EGFR and that coordinated targeting of SRC with BRAF ± EGFR increases treatment efficacy in vitro and in vivo. SRC drives resistance to BRAF ± EGFR targeted therapy independently of ERK signaling by inducing transcriptional reprogramming through β-catenin (CTNNB1). The EGFR-independent compensatory activation of SRC kinases is mediated by an autocrine prostaglandin E2 loop that can be blocked with cyclooxygenase-2 (COX2) inhibitors. Co-targeting of COX2 with BRAF + EGFR promotes durable suppression of tumor growth in patient-derived tumor xenograft models. COX2 inhibition represents a drug-repurposing strategy to overcome therapeutic resistance in BRAFV600E CRC.
© 2023. The Author(s).
Conflict of interest statement
C.E.A. served on the scientific advisory boards of Array Biopharma, Pfizer and Pionyr Immunotherapeutics and has received research funding (institutional) from Bristol Meyers Squibb, Erasca, Guardant Health, Kura Oncology, Merck and Novartis. The remaining authors declare no competing interests.
Figures








References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
