

Mitochondrial calcium cycling in neuronal function and neurodegeneration
- PMID: 36760367
- PMCID: PMC9902777
- DOI: 10.3389/fcell.2023.1094356
Mitochondrial calcium cycling in neuronal function and neurodegeneration
Abstract
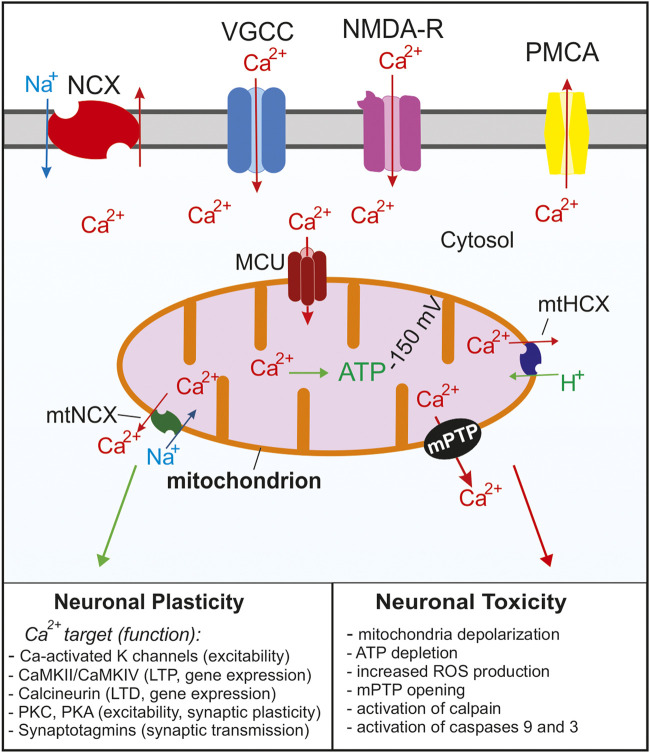
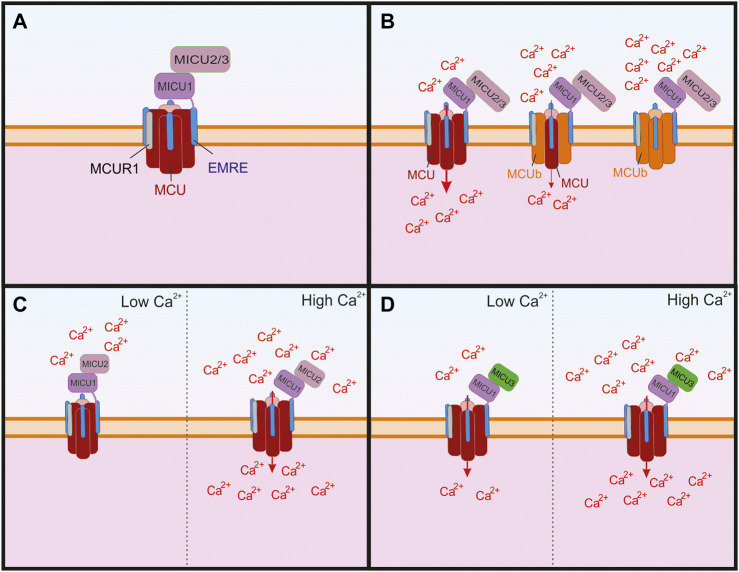
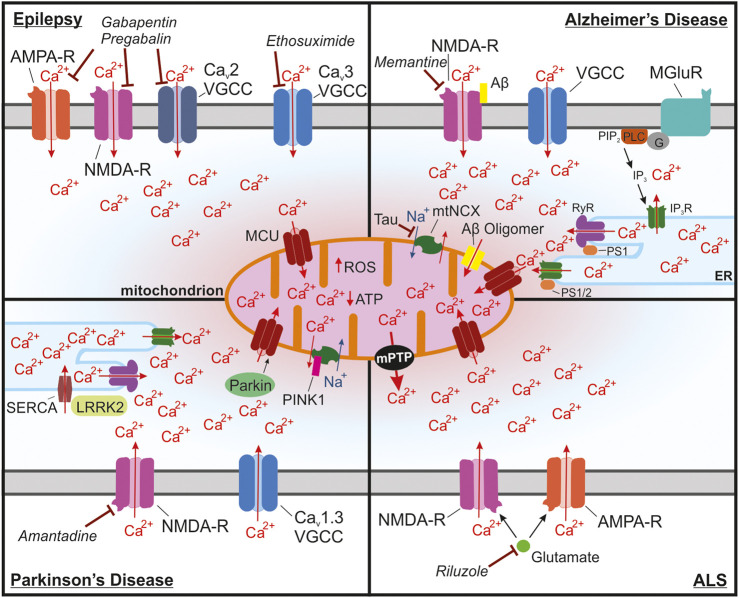
Mitochondria are essential for proper cellular function through their critical roles in ATP synthesis, reactive oxygen species production, calcium (Ca2+) buffering, and apoptotic signaling. In neurons, Ca2+ buffering is particularly important as it helps to shape Ca2+ signals and to regulate numerous Ca2+-dependent functions including neuronal excitability, synaptic transmission, gene expression, and neuronal toxicity. Over the past decade, identification of the mitochondrial Ca2+ uniporter (MCU) and other molecular components of mitochondrial Ca2+ transport has provided insight into the roles that mitochondrial Ca2+ regulation plays in neuronal function in health and disease. In this review, we discuss the many roles of mitochondrial Ca2+ uptake and release mechanisms in normal neuronal function and highlight new insights into the Ca2+-dependent mechanisms that drive mitochondrial dysfunction in neurologic diseases including epilepsy, Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. We also consider how targeting Ca2+ uptake and release mechanisms could facilitate the development of novel therapeutic strategies for neurological diseases.
Keywords: MCU; calcium; mitochondria; neurodegeneration; neuronal calcium homeostasis.
Copyright © 2023 Walters and Usachev.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Akundi R. S., Huang Z., Eason J., Pandya J. D., Zhi L., Cass W. A., et al. (2011). Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6 (1), e16038. 10.1371/journal.pone.0016038 - DOI - PMC - PubMed
-
- Alavian K. N., Beutner G., Lazrove E., Sacchetti S., Park H.-A., Licznerski P., et al. (2014). An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. 111(29), 10580–10585. 10.1073/pnas.1401591111 - DOI - PMC - PubMed
-
- Alzheimer's and Dementia (2020a). Alzheimer's disease facts and figures. Alzheimer's Dementia 16 (3), 391–460. 10.1002/alz.12068 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous