

Quinate-based ligands for irreversible inactivation of the bacterial virulence factor DHQ1 enzyme-A molecular insight
- PMID: 36762206
- PMCID: PMC9902378
- DOI: 10.3389/fmolb.2023.1111598
Quinate-based ligands for irreversible inactivation of the bacterial virulence factor DHQ1 enzyme-A molecular insight
Abstract
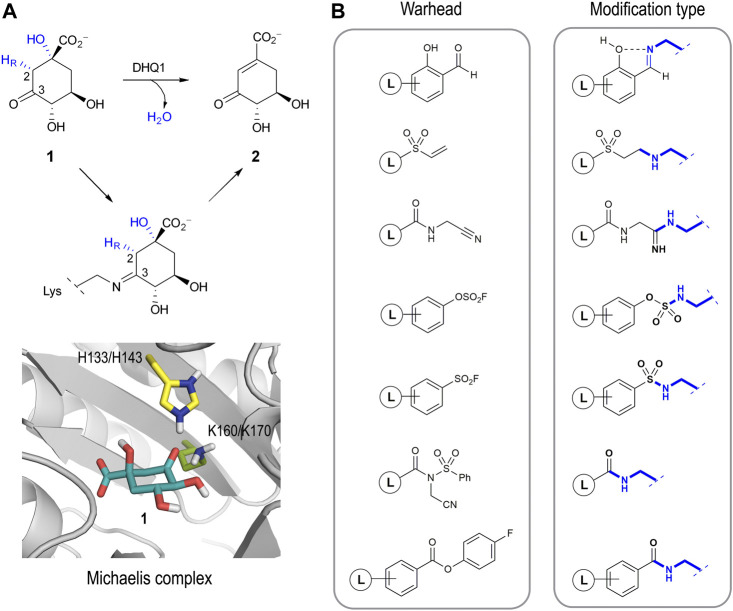
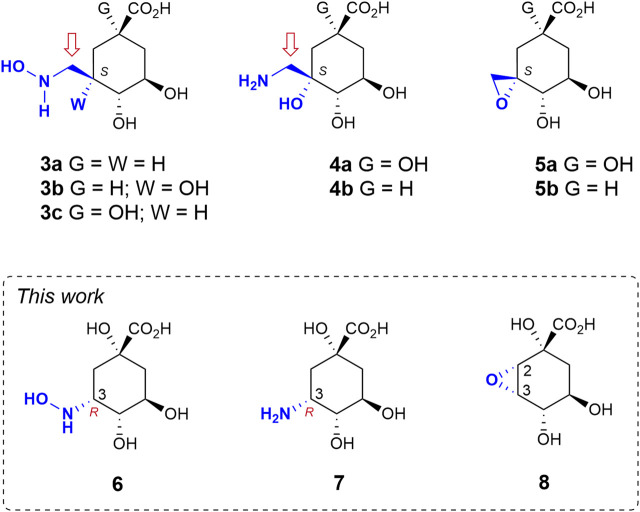
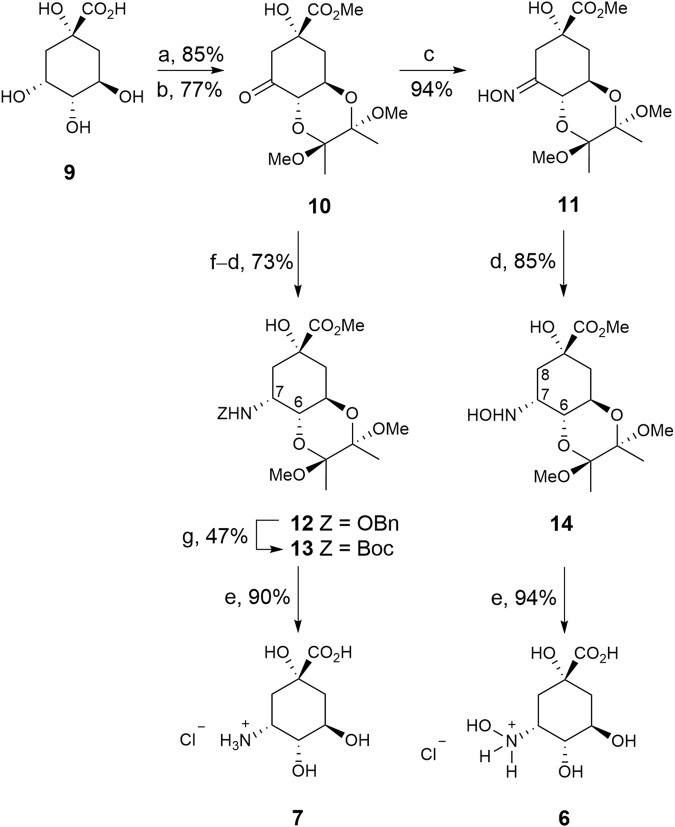
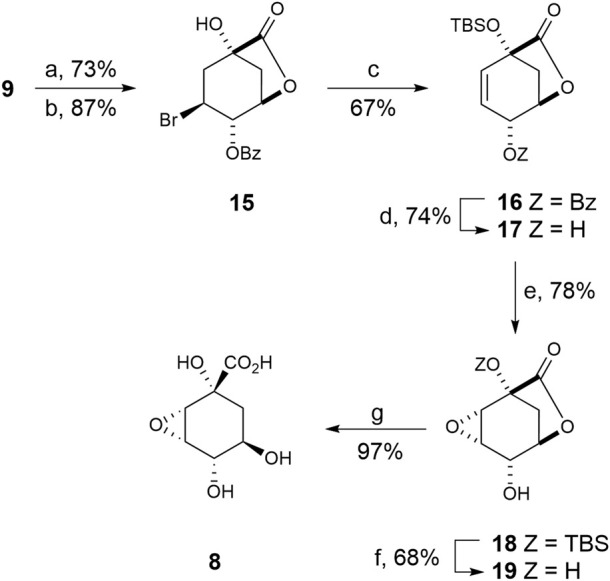
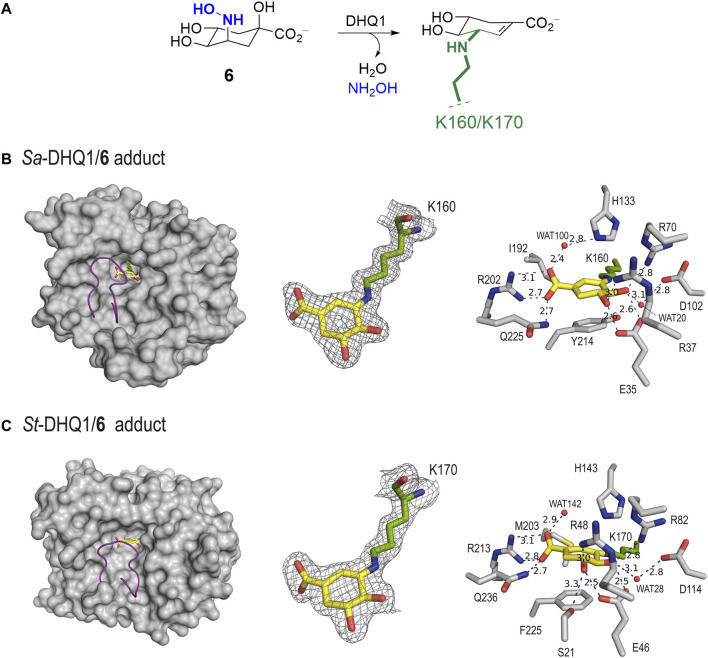
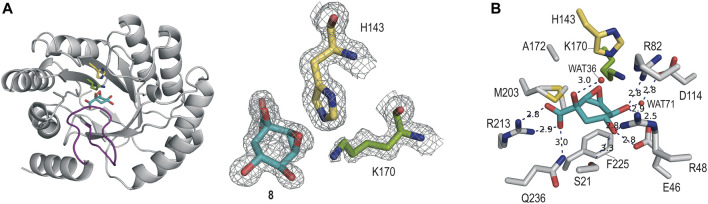
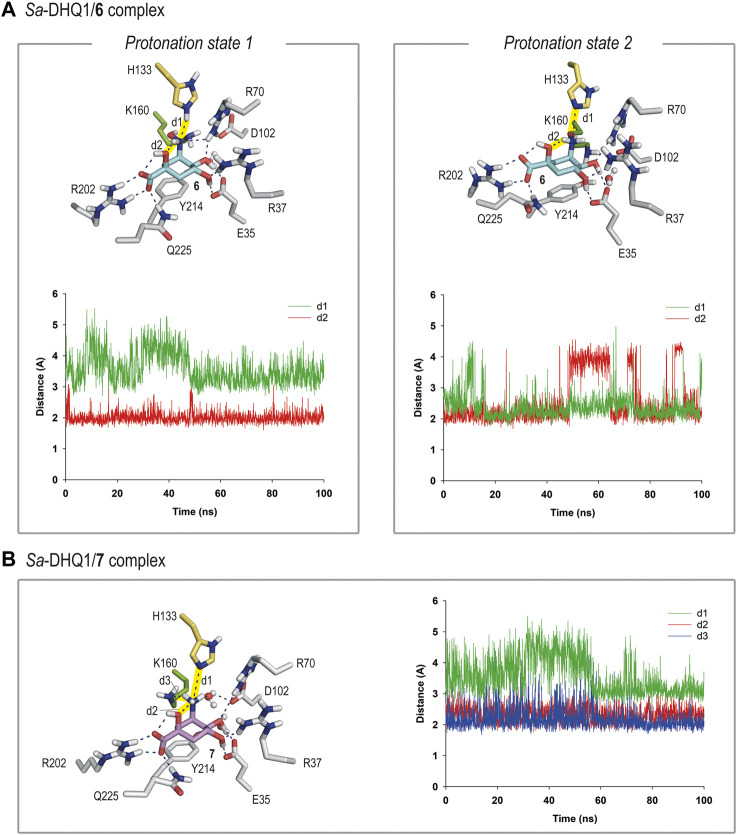
Irreversible inhibition of the enzyme type I dehydroquinase (DHQ1), a promising target for anti-virulence drug development, has been explored by enhancing the electrophilicity of specific positions of the ligand towards covalent lysine modification. For ligand design, we made use of the advantages offered by the intrinsic acid-base properties of the amino substituents introduced in the quinate scaffold, namely compounds 6-7 (R configuration at C3), to generate a potential leaving group, as well as the recognition pattern of the enzyme. The reactivity of the C2-C3 bond (Re face) in the scaffold was also explored using compound 8. The results of the present study show that replacement of the C3 hydroxy group of (-)-quinic acid by a hydroxyamino substituent (compound 6) provides a time-dependent irreversible inhibitor, while compound 7, in which the latter functionality was substituted by an amino group, and the introduction of an oxirane ring at C2-C3 bond, compound 8, do not allow covalent modification of the enzyme. These outcomes were supported by resolution of the crystal structures of DHQ1 from Staphylococcus aureus (Sa-DHQ1) and Salmonella typhi (St-DHQ1) chemically modified by 6 at a resolution of 1.65 and 1.90 Å, respectively, and of St-DHQ1 in the complex with 8 (1.55 Å). The combination of these structural studies with extensive molecular dynamics simulation studies allowed us to understand the molecular basis of the type of inhibition observed. This study is a good example of the importance of achieving the correct geometry between the reactive center of the ligand (electrophile) and the enzyme nucleophile (lysine residue) to allow selective covalent modification. The outcomes obtained with the hydroxyamino derivative 6 also open up new possibilities in the design of irreversible inhibitors based on the use of amino substituents.
Keywords: anti-virulence agents; binding mode; biomacromolecule simulations; enzyme recognition; irreversible inhibition; ligand activation; organic synthesis; type I dehydroquinase.
Copyright © 2023 Rodríguez, Maneiro, Lence, Otero, van Raaij, Thompson, Hawkins and González-Bello.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Chemical Modification of a Dehydratase Enzyme Involved in Bacterial Virulence by an Ammonium Derivative: Evidence of its Active Site Covalent Adduct.J Am Chem Soc. 2015 Jul 29;137(29):9333-43. doi: 10.1021/jacs.5b04080. Epub 2015 Jul 16. J Am Chem Soc. 2015. PMID: 26148116
-
Irreversible covalent modification of type I dehydroquinase with a stable Schiff base.Org Biomol Chem. 2015 Jan 21;13(3):706-16. doi: 10.1039/c4ob01782j. Org Biomol Chem. 2015. PMID: 25370445
-
Self-Immolation of a Bacterial Dehydratase Enzyme by its Epoxide Product.Chemistry. 2020 Jun 26;26(36):8035-8044. doi: 10.1002/chem.202000759. Epub 2020 Jun 8. Chemistry. 2020. PMID: 32259333
-
Specific chemical modification of bacterial type I dehydroquinase--opportunities for drug discovery.Future Med Chem. 2015;7(17):2371-83. doi: 10.4155/fmc.15.145. Epub 2015 Nov 24. Future Med Chem. 2015. PMID: 26599605 Review.
-
Irreversible protein kinase inhibitors.Curr Med Chem. 2011;18(20):2981-94. doi: 10.2174/092986711796391705. Curr Med Chem. 2011. PMID: 21651479 Review.
References
-
- Bartlett P. A., Maitra U., Chouinard P. M. (1986). Synthesis of "iso-EPSP" and evaluation of its interaction with chorismate synthase. J. Am. Chem. Soc. 108 (25), 8068–8071. 10.1021/ja00285a031 - DOI
-
- Boulet P., Gilardoni F., Weber J., Chermette H., Ellinger Y. (1999). Theoretical study of interstellar hydroxylamine chemistry: Protonation and proton transfer mediated by H3+. Chem. Phys. 244 (2), 163–174. 10.1016/s0301-0104(99)00151-2 - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous