

Learning high-order interactions for polygenic risk prediction
- PMID: 36763605
- PMCID: PMC9916647
- DOI: 10.1371/journal.pone.0281618
Learning high-order interactions for polygenic risk prediction
Abstract
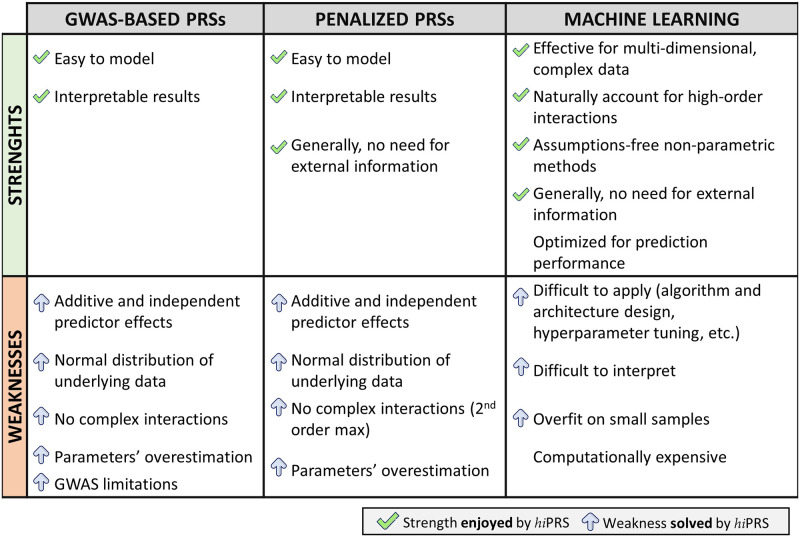
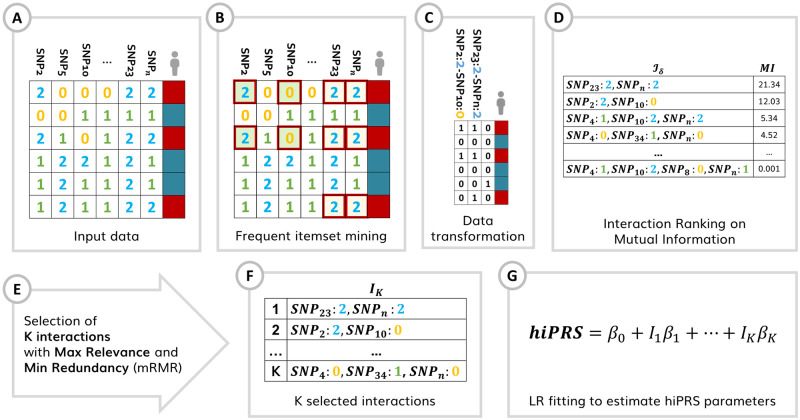
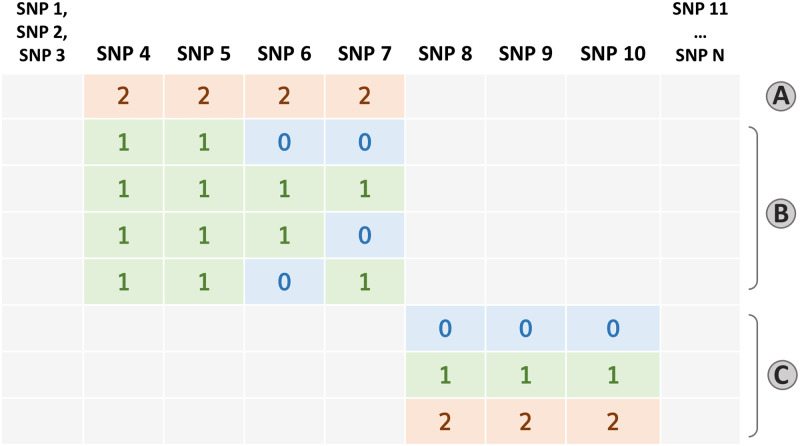
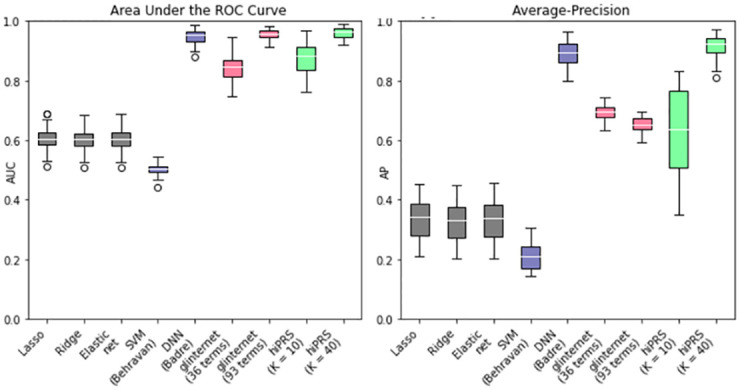
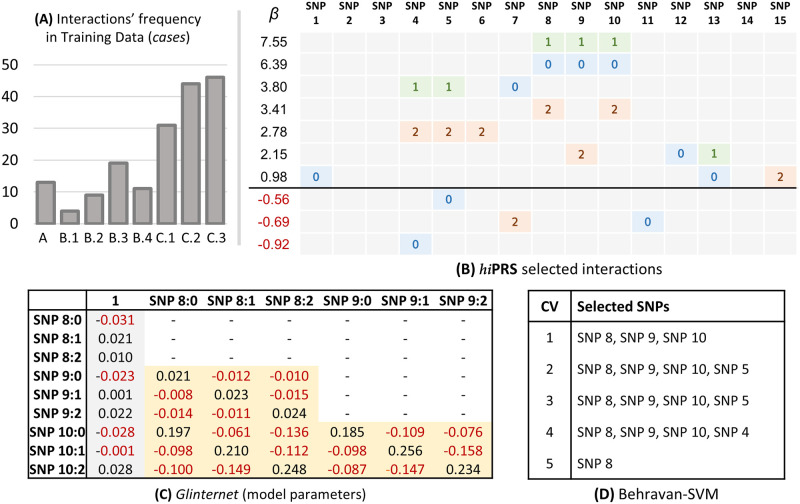
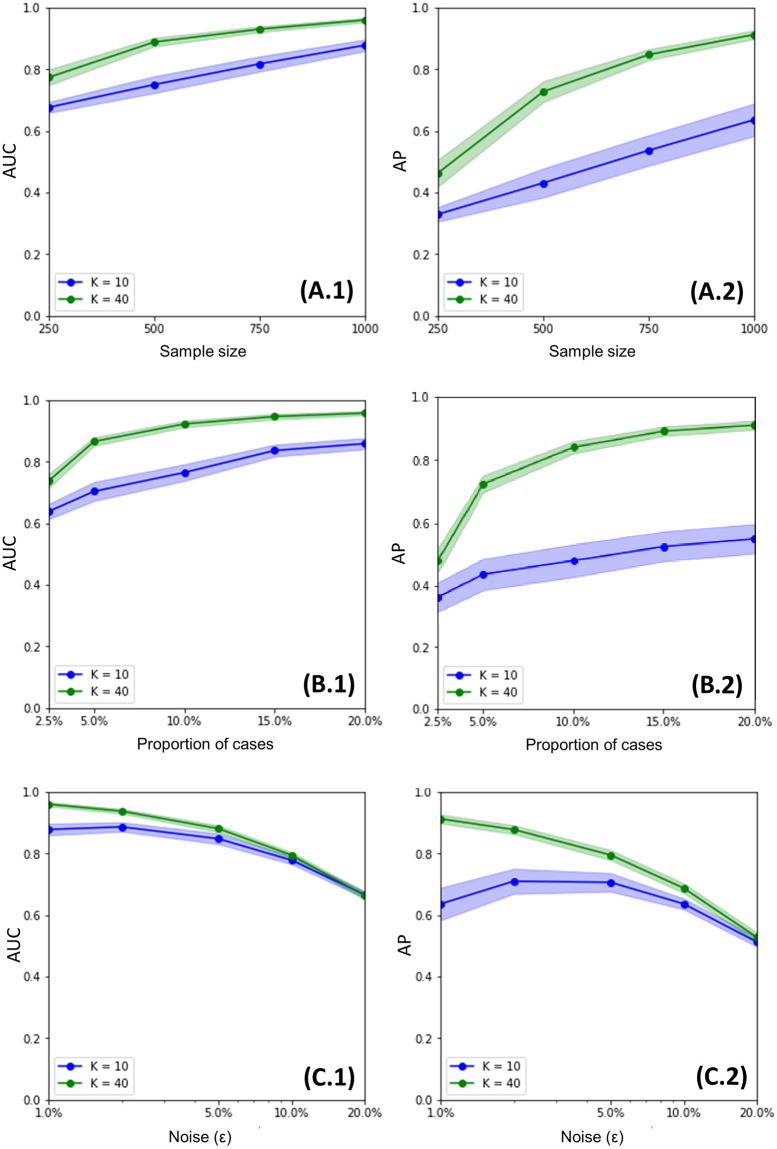
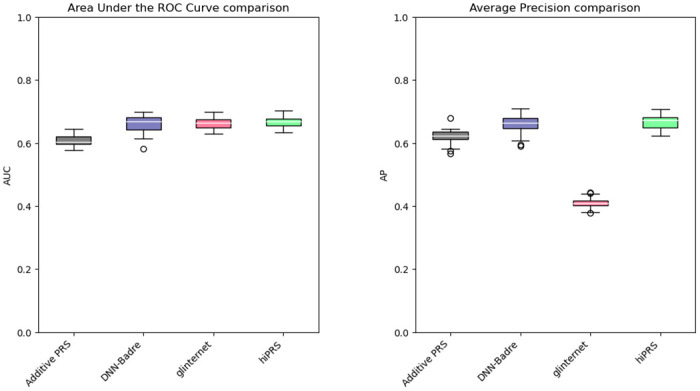
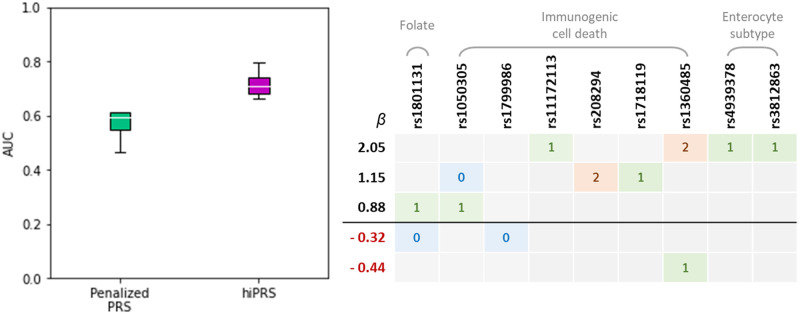
Within the framework of precision medicine, the stratification of individual genetic susceptibility based on inherited DNA variation has paramount relevance. However, one of the most relevant pitfalls of traditional Polygenic Risk Scores (PRS) approaches is their inability to model complex high-order non-linear SNP-SNP interactions and their effect on the phenotype (e.g. epistasis). Indeed, they incur in a computational challenge as the number of possible interactions grows exponentially with the number of SNPs considered, affecting the statistical reliability of the model parameters as well. In this work, we address this issue by proposing a novel PRS approach, called High-order Interactions-aware Polygenic Risk Score (hiPRS), that incorporates high-order interactions in modeling polygenic risk. The latter combines an interaction search routine based on frequent itemsets mining and a novel interaction selection algorithm based on Mutual Information, to construct a simple and interpretable weighted model of user-specified dimensionality that can predict a given binary phenotype. Compared to traditional PRSs methods, hiPRS does not rely on GWAS summary statistics nor any external information. Moreover, hiPRS differs from Machine Learning-based approaches that can include complex interactions in that it provides a readable and interpretable model and it is able to control overfitting, even on small samples. In the present work we demonstrate through a comprehensive simulation study the superior performance of hiPRS w.r.t. state of the art methods, both in terms of scoring performance and interpretability of the resulting model. We also test hiPRS against small sample size, class imbalance and the presence of noise, showcasing its robustness to extreme experimental settings. Finally, we apply hiPRS to a case study on real data from DACHS cohort, defining an interaction-aware scoring model to predict mortality of stage II-III Colon-Rectal Cancer patients treated with oxaliplatin.
Copyright: © 2023 Massi et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Epistatic Features and Machine Learning Improve Alzheimer's Disease Risk Prediction Over Polygenic Risk Scores.J Alzheimers Dis. 2024;99(4):1425-1440. doi: 10.3233/JAD-230236. J Alzheimers Dis. 2024. PMID: 38788065 Free PMC article.
-
Improving the Utility of Polygenic Risk Scores as a Biomarker for Alzheimer's Disease.Cells. 2021 Jun 29;10(7):1627. doi: 10.3390/cells10071627. Cells. 2021. PMID: 34209762 Free PMC article.
-
netCRS: Network-based comorbidity risk score for prediction of myocardial infarction using biobank-scaled PheWAS data.Pac Symp Biocomput. 2022;27:325-336. Pac Symp Biocomput. 2022. PMID: 34890160 Free PMC article.
-
Validity of polygenic risk scores: are we measuring what we think we are?Hum Mol Genet. 2019 Nov 21;28(R2):R143-R150. doi: 10.1093/hmg/ddz205. Hum Mol Genet. 2019. PMID: 31504522 Free PMC article. Review.
-
Polygenic risk for schizophrenia and associated brain structural changes: A systematic review.Compr Psychiatry. 2019 Jan;88:77-82. doi: 10.1016/j.comppsych.2018.11.014. Epub 2018 Nov 29. Compr Psychiatry. 2019. PMID: 30529765
Cited by
-
A perspective on genetic and polygenic risk scores-advances and limitations and overview of associated tools.Brief Bioinform. 2024 Mar 27;25(3):bbae240. doi: 10.1093/bib/bbae240. Brief Bioinform. 2024. PMID: 38770718 Free PMC article. Review.
-
Epistatic Features and Machine Learning Improve Alzheimer's Disease Risk Prediction Over Polygenic Risk Scores.J Alzheimers Dis. 2024;99(4):1425-1440. doi: 10.3233/JAD-230236. J Alzheimers Dis. 2024. PMID: 38788065 Free PMC article.
-
Machine Learning Models of Polygenic Risk for Enhanced Prediction of Alzheimer Disease Endophenotypes.Neurol Genet. 2024 Jan 10;10(1):e200120. doi: 10.1212/NXG.0000000000200120. eCollection 2024 Feb. Neurol Genet. 2024. PMID: 38250184 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
