

Flipping Off and On the Redox Switch in the Microcirculation
- PMID: 36763969
- PMCID: PMC11046419
- DOI: 10.1146/annurev-physiol-031522-021457
Flipping Off and On the Redox Switch in the Microcirculation
Abstract
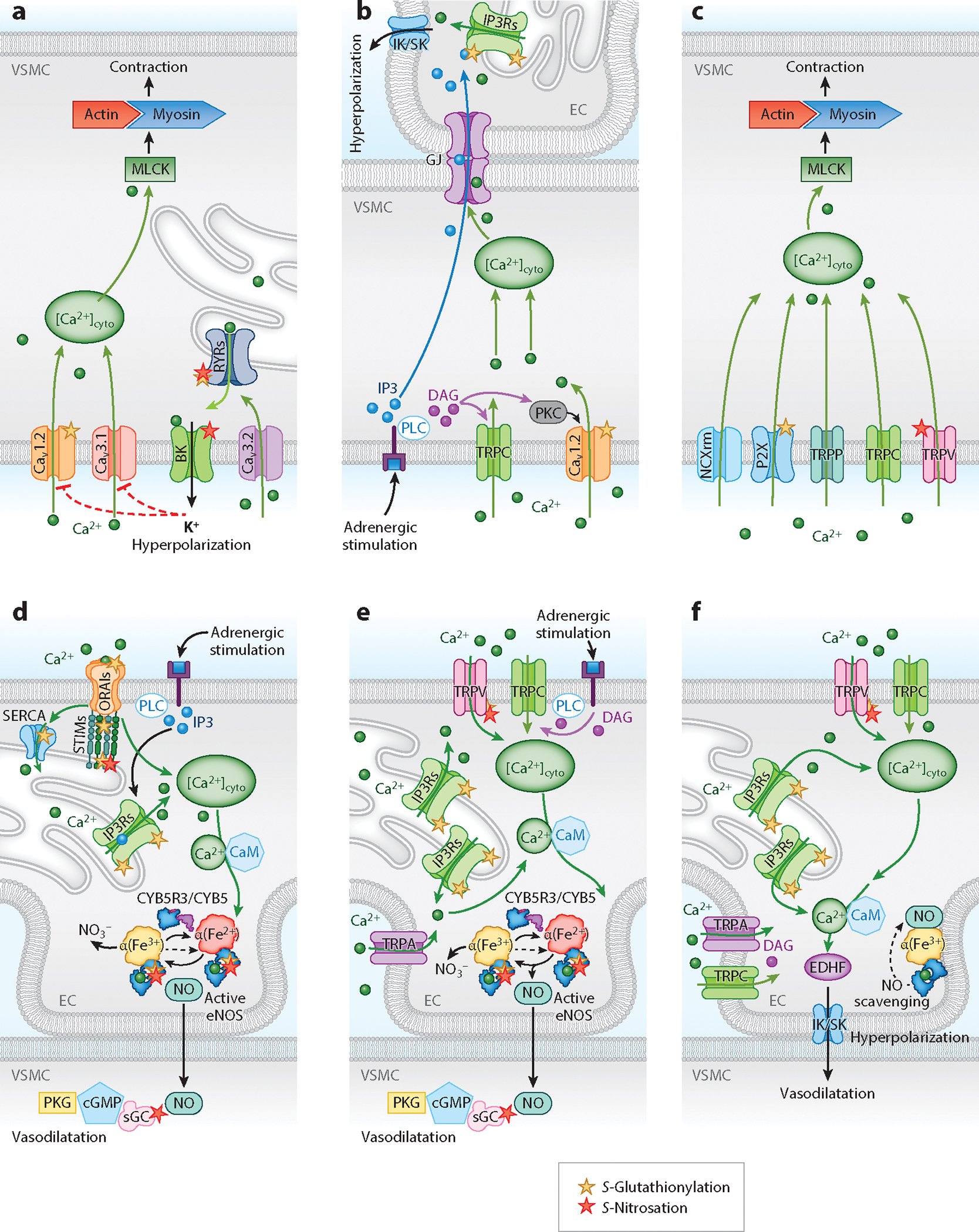
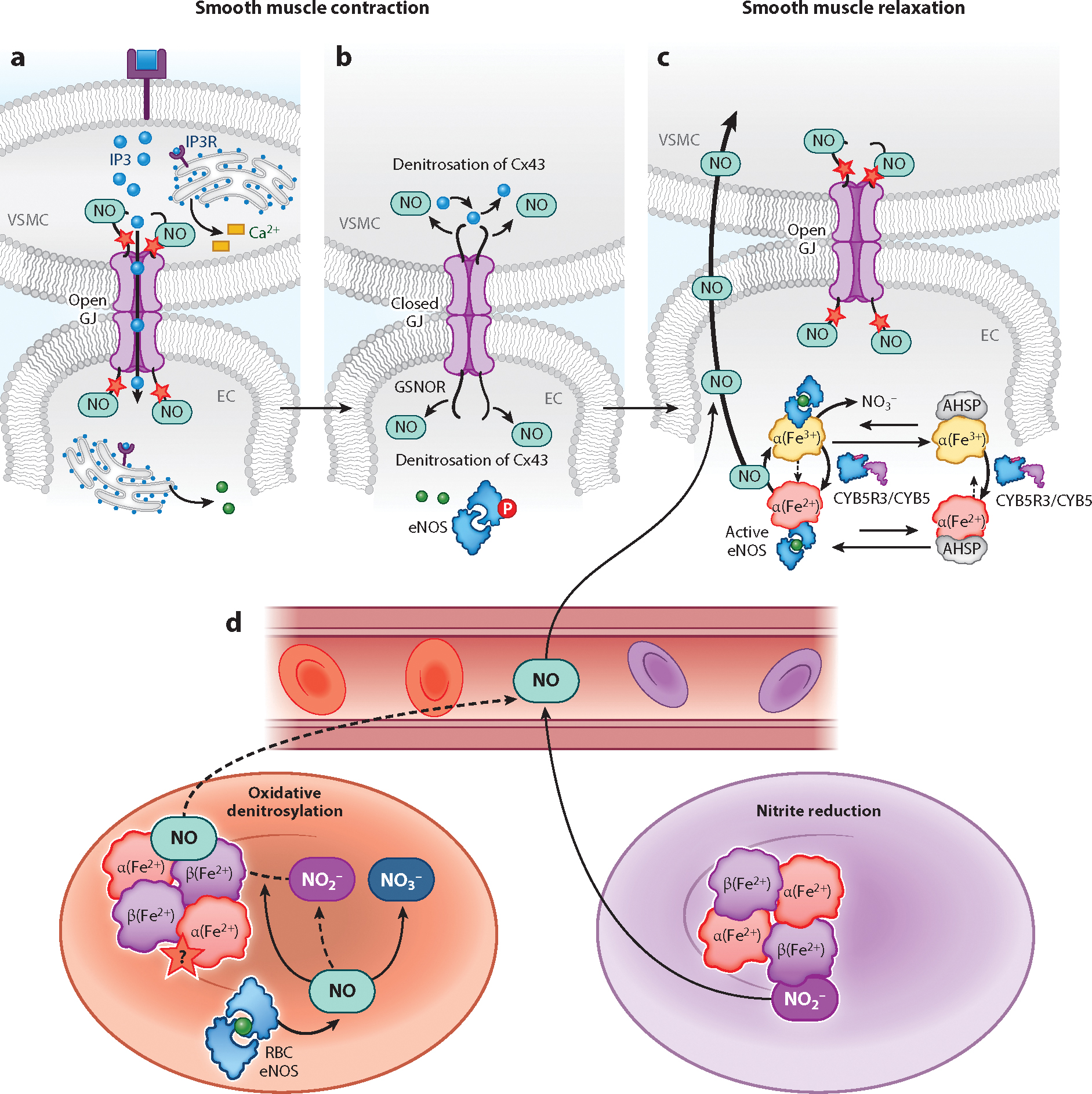
Resistance arteries and arterioles evolved as specialized blood vessels serving two important functions: (a) regulating peripheral vascular resistance and blood pressure and (b) matching oxygen and nutrient delivery to metabolic demands of organs. These functions require control of vessel lumen cross-sectional area (vascular tone) via coordinated vascular cell responses governed by precise spatial-temporal communication between intracellular signaling pathways. Herein, we provide a contemporary overview of the significant roles that redox switches play in calcium signaling for orchestrated endothelial, smooth muscle, and red blood cell control of arterial vascular tone. Three interrelated themes are the focus: (a) smooth muscle to endothelial communication for vasoconstriction, (b) endothelial to smooth muscle cell cross talk for vasodilation, and (c) oxygen and red blood cell interregulation of vascular tone and blood flow. We intend for this thematic framework to highlight gaps in our current knowledge and potentially spark interest for cross-disciplinary studies moving forward.
Keywords: calcium; endothelial; microcirculation; red blood cell; redox; smooth muscle.
Figures
References
-
- Rhodin JA. 1967. The ultrastructure of mammalian arterioles and precapillary sphincters. J. Ultrastruct. Res. 18:181–223 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
