

MEK1 drives oncogenic signaling and interacts with PARP1 for genomic and metabolic homeostasis in malignant pleural mesothelioma
- PMID: 36765038
- PMCID: PMC9918536
- DOI: 10.1038/s41420-023-01307-2
MEK1 drives oncogenic signaling and interacts with PARP1 for genomic and metabolic homeostasis in malignant pleural mesothelioma
Abstract
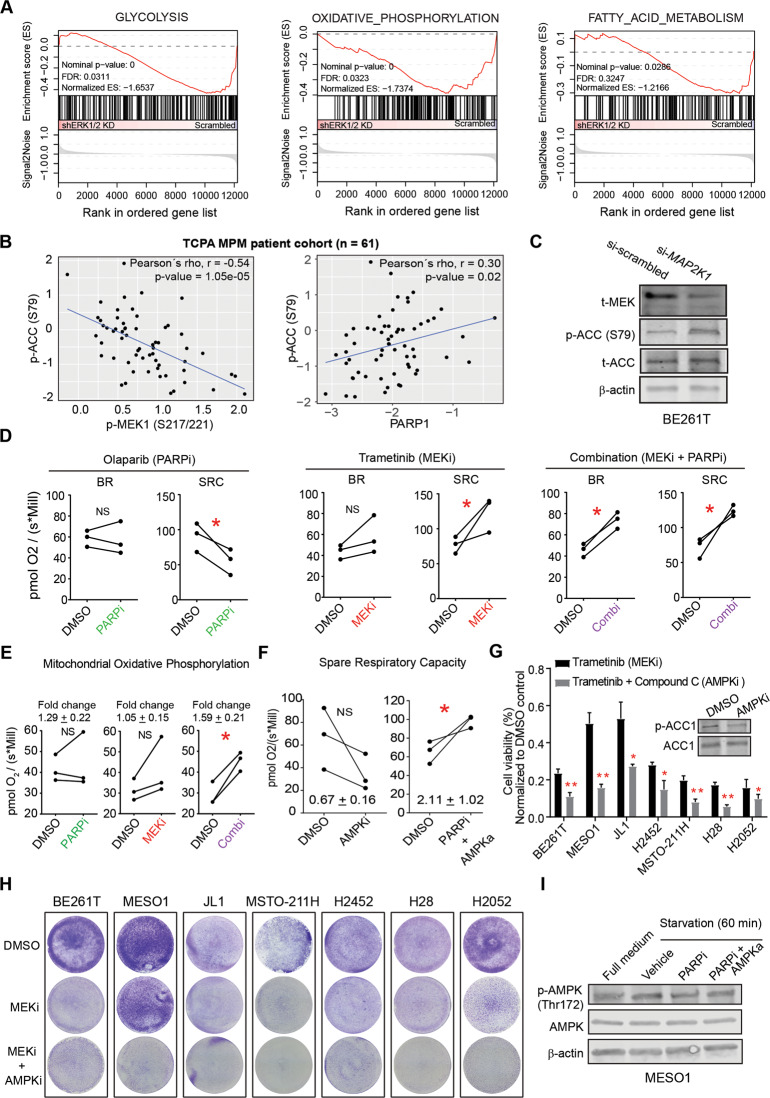
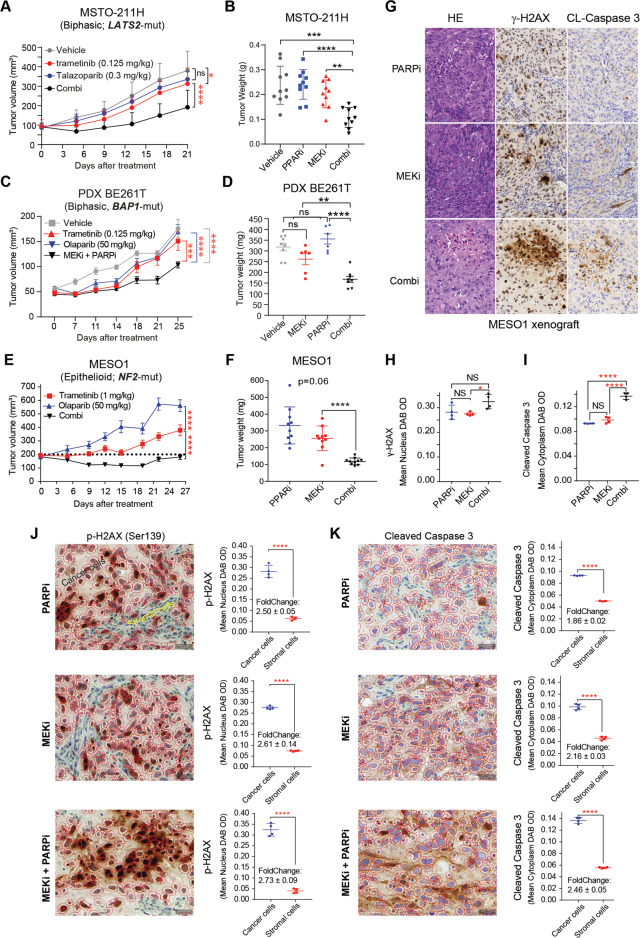
Malignant pleural mesothelioma (MPM) is a lethal malignancy etiologically caused by asbestos exposure, for which there are few effective treatment options. Although asbestos carcinogenesis is associated with reactive oxygen species (ROS), the bona fide oncogenic signaling pathways that regulate ROS homeostasis and bypass ROS-evoked apoptosis in MPM are poorly understood. In this study, we demonstrate that the mitogen-activated protein kinase (MAPK) pathway RAS-RAF-MEK-ERK is hyperactive and a molecular driver of MPM, independent of histological subtypes and genetic heterogeneity. Suppression of MAPK signaling by clinically approved MEK inhibitors (MEKi) elicits PARP1 to protect MPM cells from the cytotoxic effects of MAPK pathway blockage. Mechanistically, MEKi induces impairment of homologous recombination (HR) repair proficiency and mitochondrial metabolic activity, which is counterbalanced by pleiotropic PARP1. Consequently, the combination of MEK with PARP inhibitors enhances apoptotic cell death in vitro and in vivo that occurs through coordinated upregulation of cytotoxic ROS in MPM cells, suggesting a mechanism-based, readily translatable strategy to treat this daunting disease. Collectively, our studies uncover a previously unrecognized scenario that hyperactivation of the MAPK pathway is an essential feature of MPM and provide unprecedented evidence that MAPK signaling cooperates with PARP1 to homeostatically maintain ROS levels and escape ROS-mediated apoptosis.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Janes SM, Alrifai D, Fennell DA. Perspectives on the treatment of malignant pleural mesothelioma. New Engl J Med. 2021;385:1207–18. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
