

Bi-allelic ATG4D variants are associated with a neurodevelopmental disorder characterized by speech and motor impairment
- PMID: 36765070
- PMCID: PMC9918471
- DOI: 10.1038/s41525-022-00343-8
Bi-allelic ATG4D variants are associated with a neurodevelopmental disorder characterized by speech and motor impairment
Abstract
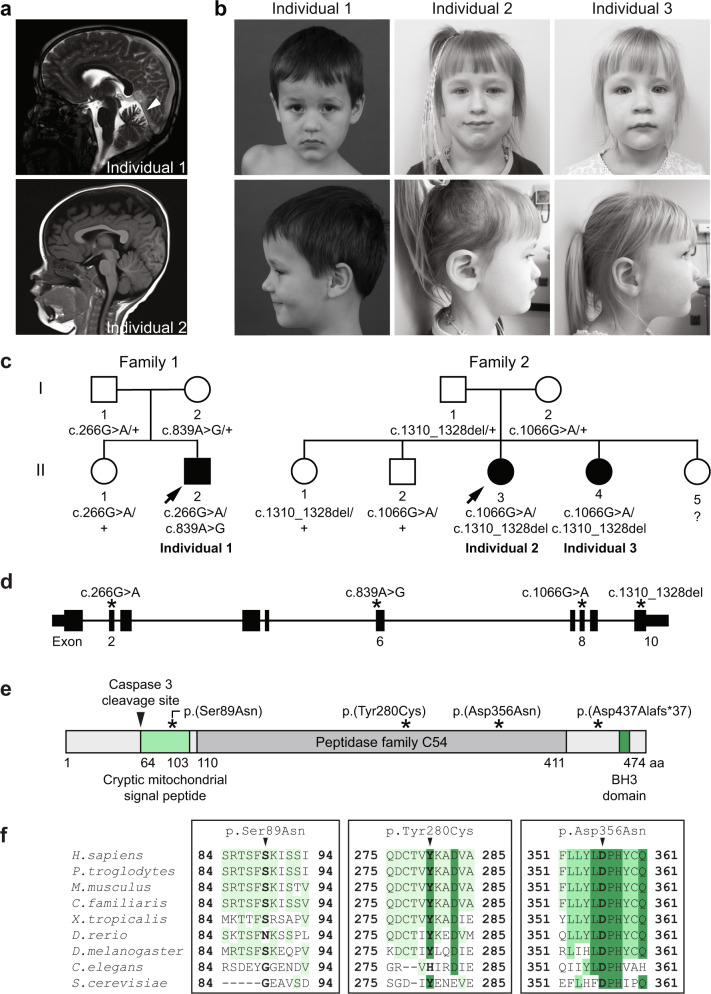
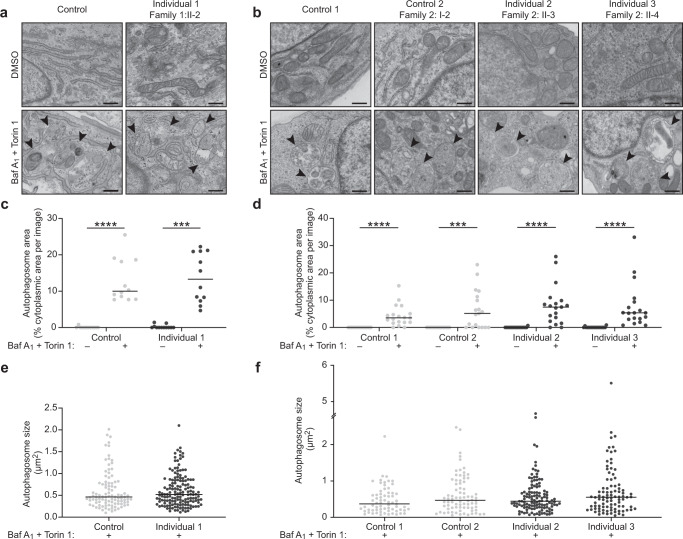
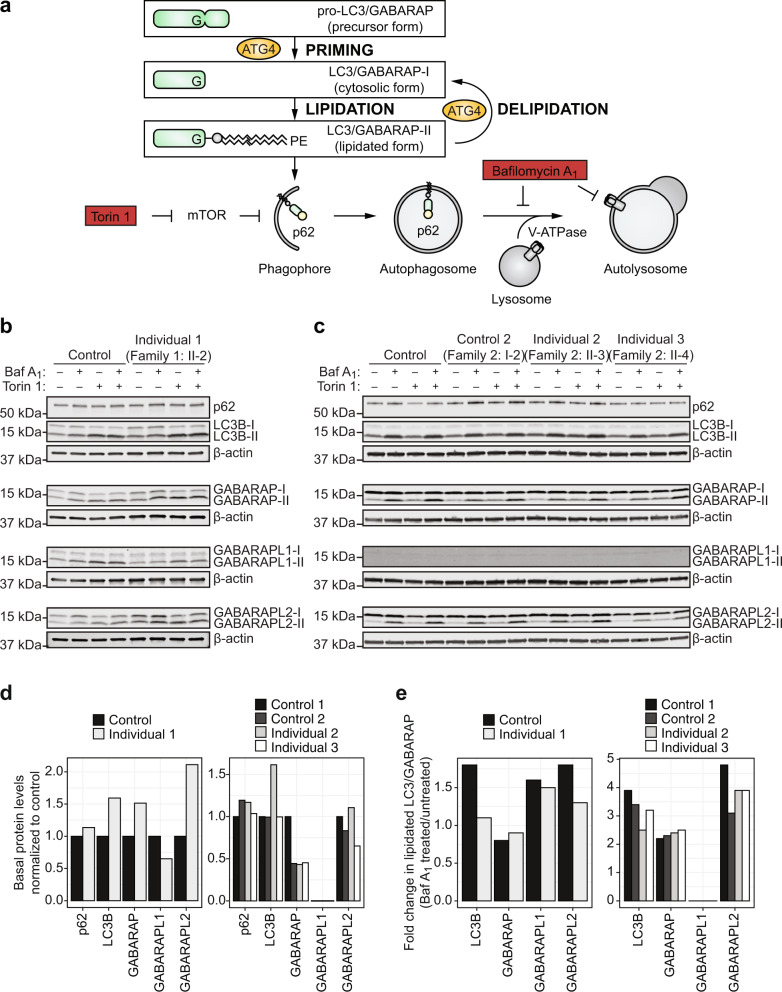
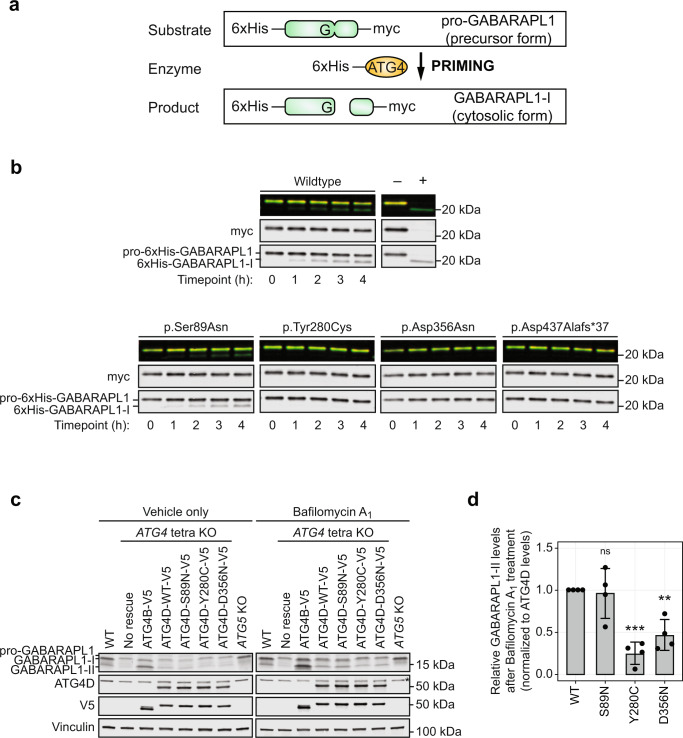
Autophagy regulates the degradation of damaged organelles and protein aggregates, and is critical for neuronal development, homeostasis, and maintenance, yet few neurodevelopmental disorders have been associated with pathogenic variants in genes encoding autophagy-related proteins. We report three individuals from two unrelated families with a neurodevelopmental disorder characterized by speech and motor impairment, and similar facial characteristics. Rare, conserved, bi-allelic variants were identified in ATG4D, encoding one of four ATG4 cysteine proteases important for autophagosome biogenesis, a hallmark of autophagy. Autophagosome biogenesis and induction of autophagy were intact in cells from affected individuals. However, studies evaluating the predominant substrate of ATG4D, GABARAPL1, demonstrated that three of the four ATG4D patient variants functionally impair ATG4D activity. GABARAPL1 is cleaved or "primed" by ATG4D and an in vitro GABARAPL1 priming assay revealed decreased priming activity for three of the four ATG4D variants. Furthermore, a rescue experiment performed in an ATG4 tetra knockout cell line, in which all four ATG4 isoforms were knocked out by gene editing, showed decreased GABARAPL1 priming activity for the two ATG4D missense variants located in the cysteine protease domain required for priming, suggesting that these variants impair the function of ATG4D. The clinical, bioinformatic, and functional data suggest that bi-allelic loss-of-function variants in ATG4D contribute to the pathogenesis of this syndromic neurodevelopmental disorder.
© 2023. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
