

SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation
- PMID: 36765386
- PMCID: PMC9912651
- DOI: 10.1186/s40246-023-00451-1
SpliceAI-visual: a free online tool to improve SpliceAI splicing variant interpretation
Abstract
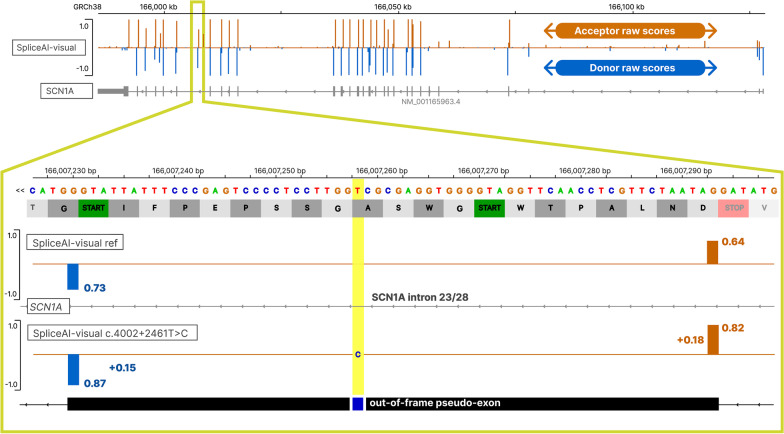
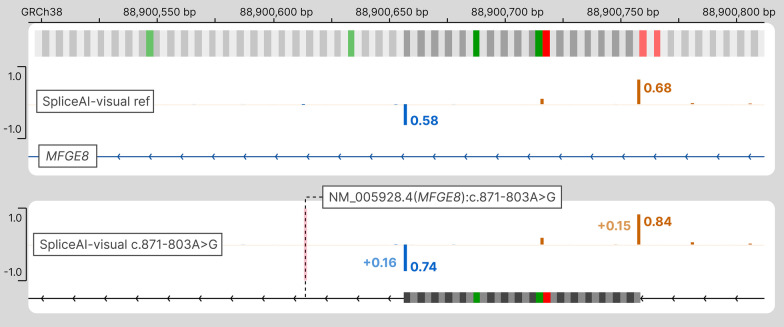
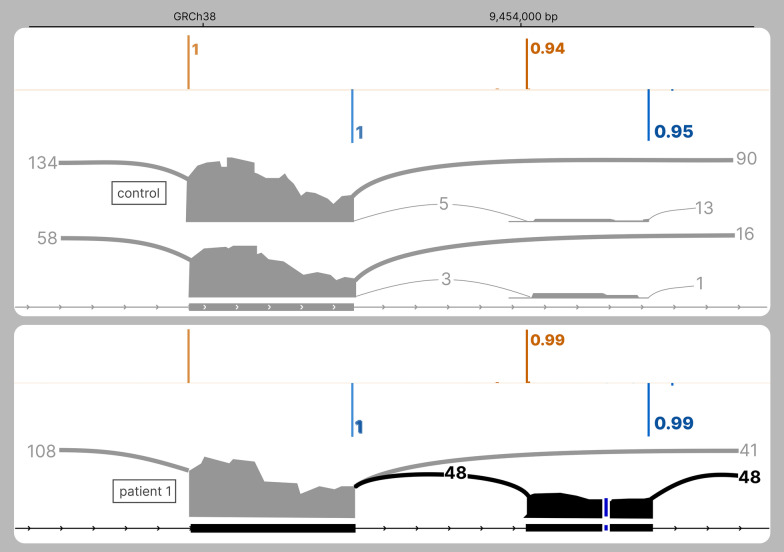
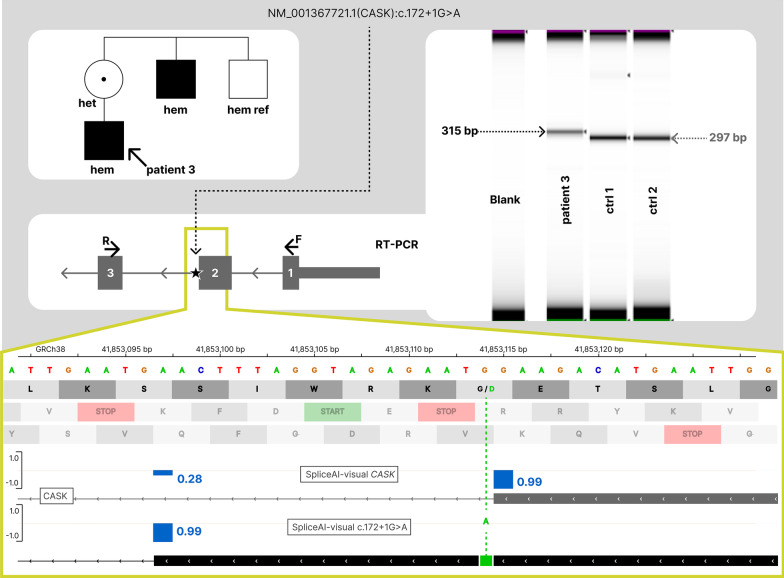
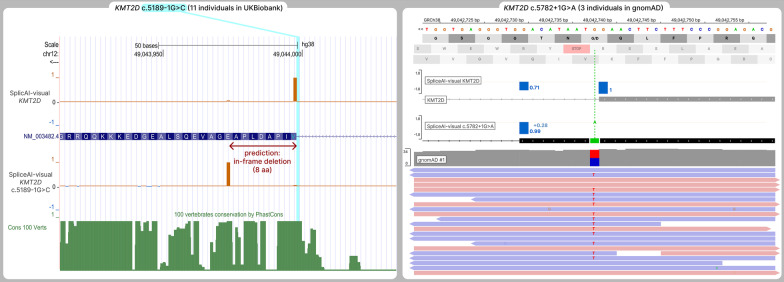
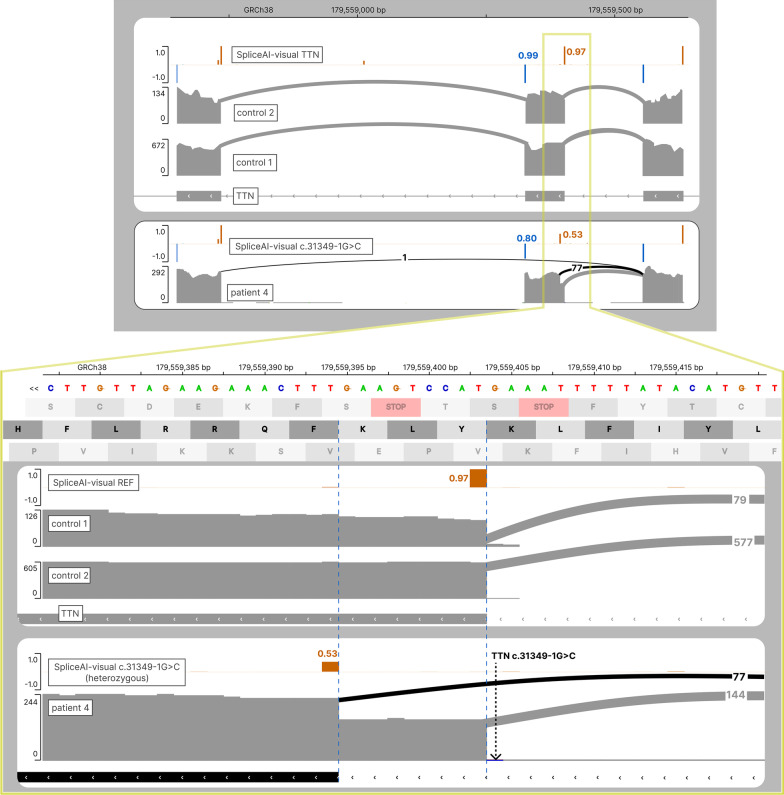
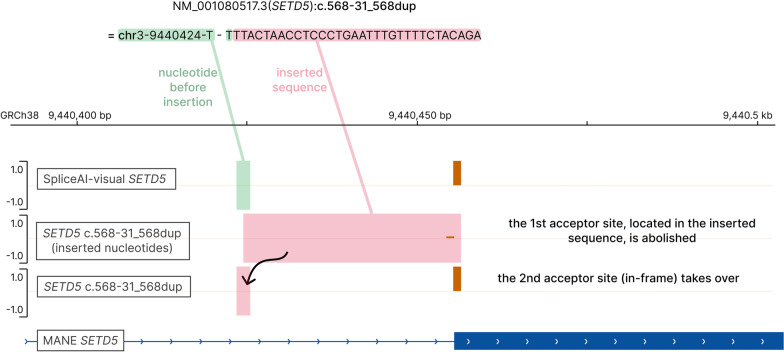
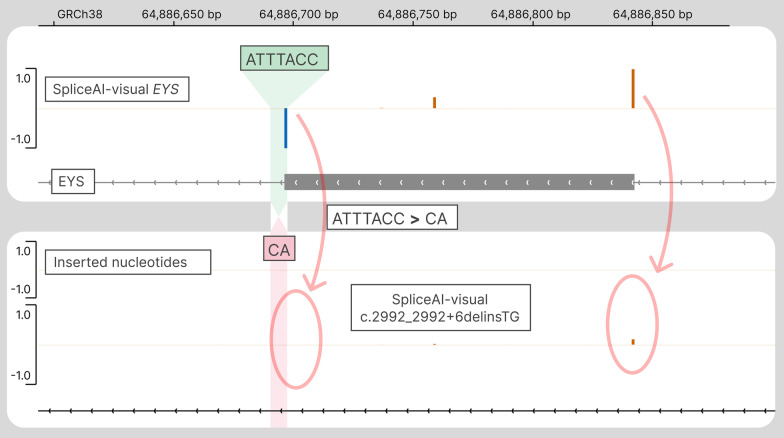
SpliceAI is an open-source deep learning splicing prediction algorithm that has demonstrated in the past few years its high ability to predict splicing defects caused by DNA variations. However, its outputs present several drawbacks: (1) although the numerical values are very convenient for batch filtering, their precise interpretation can be difficult, (2) the outputs are delta scores which can sometimes mask a severe consequence, and (3) complex delins are most often not handled. We present here SpliceAI-visual, a free online tool based on the SpliceAI algorithm, and show how it complements the traditional SpliceAI analysis. First, SpliceAI-visual manipulates raw scores and not delta scores, as the latter can be misleading in certain circumstances. Second, the outcome of SpliceAI-visual is user-friendly thanks to the graphical presentation. Third, SpliceAI-visual is currently one of the only SpliceAI-derived implementations able to annotate complex variants (e.g., complex delins). We report here the benefits of using SpliceAI-visual and demonstrate its relevance in the assessment/modulation of the PVS1 classification criteria. We also show how SpliceAI-visual can elucidate several complex splicing defects taken from the literature but also from unpublished cases. SpliceAI-visual is available as a Google Colab notebook and has also been fully integrated in a free online variant interpretation tool, MobiDetails ( https://mobidetails.iurc.montp.inserm.fr/MD ).
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
