

The RSPH4A Gene in Primary Ciliary Dyskinesia
- PMID: 36768259
- PMCID: PMC9915723
- DOI: 10.3390/ijms24031936
The RSPH4A Gene in Primary Ciliary Dyskinesia
Abstract
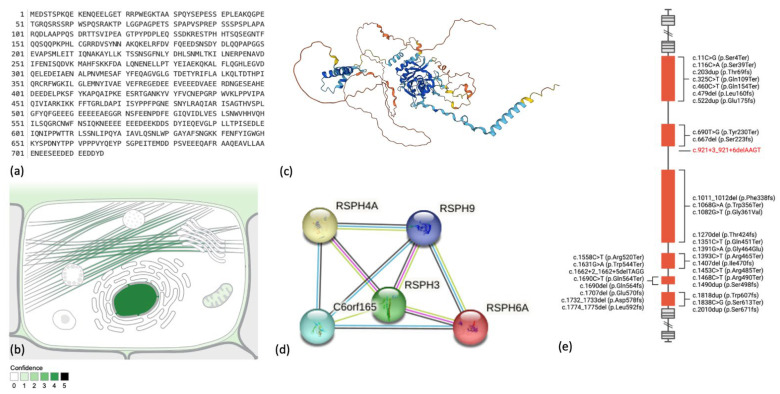
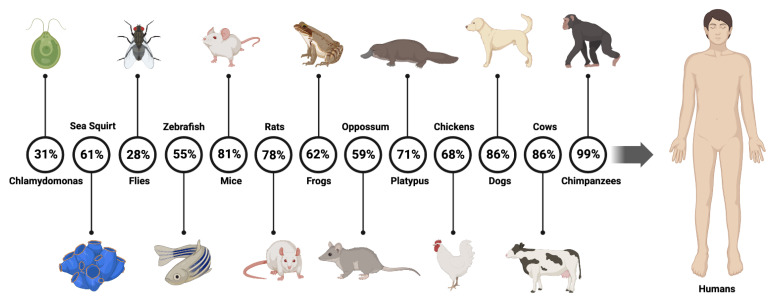
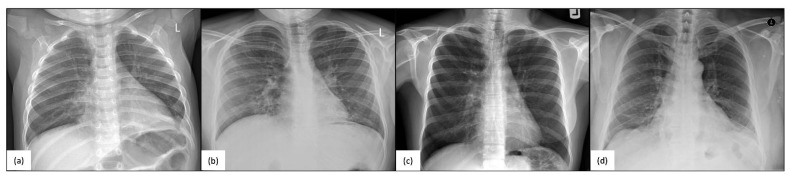
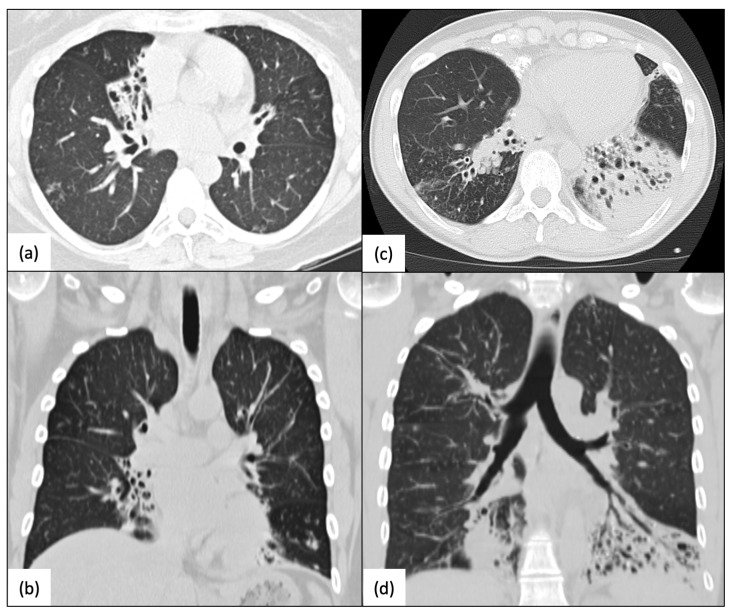
The radial spoke head protein 4 homolog A (RSPH4A) gene is one of more than 50 genes that cause Primary ciliary dyskinesia (PCD), a rare genetic ciliopathy. Genetic mutations in the RSPH4A gene alter an important protein structure involved in ciliary pathogenesis. Radial spoke proteins, such as RSPH4A, have been conserved across multiple species. In humans, ciliary function deficiency caused by RSPH4A pathogenic variants results in a clinical phenotype characterized by recurrent oto-sino-pulmonary infections. More than 30 pathogenic RSPH4A genetic variants have been associated with PCD. In Puerto Rican Hispanics, a founder mutation (RSPH4A (c.921+3_921+6delAAGT (intronic)) has been described. The spectrum of the RSPH4A PCD phenotype does not include laterality defects, which results in a challenging diagnosis. PCD diagnostic tools can combine transmission electron microscopy (TEM), nasal nitric oxide (nNO), High-Speed Video microscopy Analysis (HSVA), and immunofluorescence. The purpose of this review article is to provide a comprehensive overview of current knowledge about the RSPH4A gene in PCD, ranging from basic science to human clinical phenotype.
Keywords: RSPH4A; cilia; genotype-phenotype relationships; high-speed video microscopy analysis; immunofluorescence; nasal nitric oxide; primary ciliary dyskinesia; transmission electron microscopy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Primary Ciliary Dyskinesia Diagnostic Challenges: Understanding the Clinical Phenotype of the Puerto Rican RSPH4A Founder Mutation.Diagnostics (Basel). 2021 Feb 11;11(2):281. doi: 10.3390/diagnostics11020281. Diagnostics (Basel). 2021. PMID: 33670432 Free PMC article.
-
Immunofluorescence Analysis and Diagnosis of Primary Ciliary Dyskinesia with Radial Spoke Defects.Am J Respir Cell Mol Biol. 2015 Oct;53(4):563-73. doi: 10.1165/rcmb.2014-0483OC. Am J Respir Cell Mol Biol. 2015. PMID: 25789548 Free PMC article.
-
Founder mutation in RSPH4A identified in patients of Hispanic descent with primary ciliary dyskinesia.Hum Mutat. 2013 Oct;34(10):1352-6. doi: 10.1002/humu.22371. Epub 2013 Aug 6. Hum Mutat. 2013. PMID: 23798057 Free PMC article.
-
Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non-diagnostic ciliary ultrastructure.Ultrastruct Pathol. 2017 Nov-Dec;41(6):373-385. doi: 10.1080/01913123.2017.1362088. Epub 2017 Sep 15. Ultrastruct Pathol. 2017. PMID: 28915070 Free PMC article. Review.
-
Primary Ciliary Dyskinesia: A Clinical Review.Cells. 2024 Jun 4;13(11):974. doi: 10.3390/cells13110974. Cells. 2024. PMID: 38891105 Free PMC article. Review.
Cited by
-
Domiciliary High-Flow Nasal Therapy in Primary Ciliary Dyskinesia.Cureus. 2023 Jan 25;15(1):e34177. doi: 10.7759/cureus.34177. eCollection 2023 Jan. Cureus. 2023. PMID: 36843741 Free PMC article.
-
The underlying molecular mechanism of ciliated epithelium dysfunction and TGF-β signaling in children with congenital pulmonary airway malformations.Sci Rep. 2024 Feb 23;14(1):4430. doi: 10.1038/s41598-024-54924-x. Sci Rep. 2024. PMID: 38396057 Free PMC article.
-
Assessing Olfactory Acuity in Primary Ciliary Dyskinesia with the RSPH4A Founder Mutation.J Clin Med. 2025 May 21;14(10):3612. doi: 10.3390/jcm14103612. J Clin Med. 2025. PMID: 40429607 Free PMC article.
-
Evaluation of Open-Source Ciliary Analysis Software in Primary Ciliary Dyskinesia: A Comparative Assessment.Diagnostics (Basel). 2024 Aug 20;14(16):1814. doi: 10.3390/diagnostics14161814. Diagnostics (Basel). 2024. PMID: 39202302 Free PMC article.
-
A Pilot Study of Primary Ciliary Dyskinesia: Sleep-Related Disorders and Neuropsychiatric Comorbidities.J Clin Med. 2025 Feb 18;14(4):1353. doi: 10.3390/jcm14041353. J Clin Med. 2025. PMID: 40004884 Free PMC article.
References
-
- Leigh M.W., Ferkol T.W., Davis S.D., Lee H.-S., Rosenfeld M., Dell S.D., Sagel S.D., Milla C., Olivier K.N., Sullivan K.M., et al. Clinical Features and Associated Likelihood of Primary Ciliary Dyskinesia in Children and Adolescents. Ann. Am. Thorac. Soc. 2016;13:1305–1313. doi: 10.1513/AnnalsATS.201511-748OC. - DOI - PMC - PubMed
-
- Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C., et al. Mutations in Radial Spoke Head Protein Genes RSPH9 and RSPH4A Cause Primary Ciliary Dyskinesia with Central-Microtubular-Pair Abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
