

Molecular Mechanisms Underlying Neuroinflammation Elicited by Occupational Injuries and Toxicants
- PMID: 36768596
- PMCID: PMC9917383
- DOI: 10.3390/ijms24032272
Molecular Mechanisms Underlying Neuroinflammation Elicited by Occupational Injuries and Toxicants
Abstract
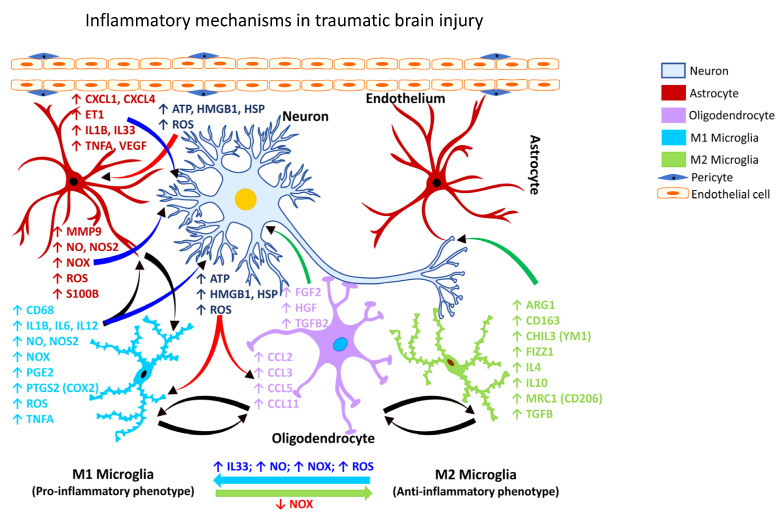
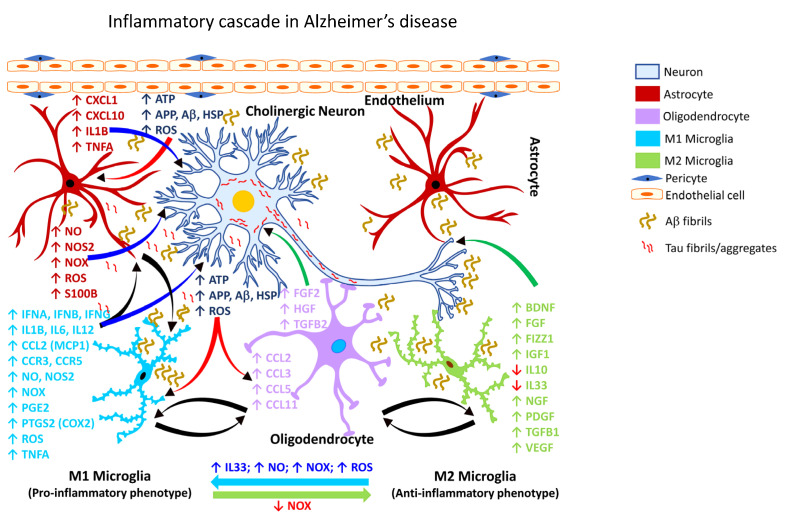
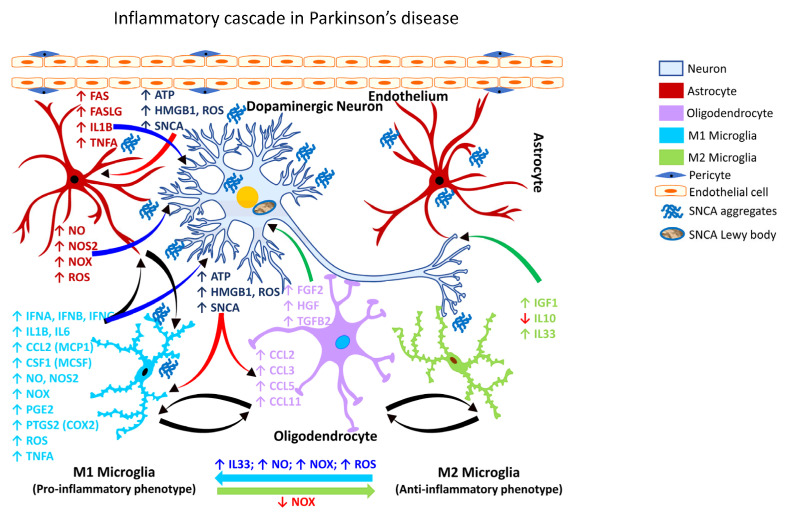
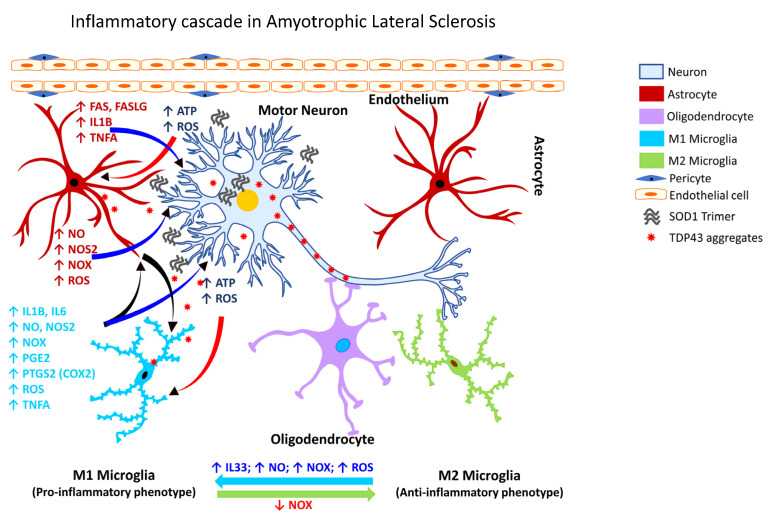
Occupational injuries and toxicant exposures lead to the development of neuroinflammation by activating distinct mechanistic signaling cascades that ultimately culminate in the disruption of neuronal function leading to neurological and neurodegenerative disorders. The entry of toxicants into the brain causes the subsequent activation of glial cells, a response known as 'reactive gliosis'. Reactive glial cells secrete a wide variety of signaling molecules in response to neuronal perturbations and thus play a crucial role in the progression and regulation of central nervous system (CNS) injury. In parallel, the roles of protein phosphorylation and cell signaling in eliciting neuroinflammation are evolving. However, there is limited understanding of the molecular underpinnings associated with toxicant- or occupational injury-mediated neuroinflammation, gliosis, and neurological outcomes. The activation of signaling molecules has biological significance, including the promotion or inhibition of disease mechanisms. Nevertheless, the regulatory mechanisms of synergism or antagonism among intracellular signaling pathways remain elusive. This review highlights the research focusing on the direct interaction between the immune system and the toxicant- or occupational injury-induced gliosis. Specifically, the role of occupational injuries, e.g., trips, slips, and falls resulting in traumatic brain injury, and occupational toxicants, e.g., volatile organic compounds, metals, and nanoparticles/nanomaterials in the development of neuroinflammation and neurological or neurodegenerative diseases are highlighted. Further, this review recapitulates the recent advancement related to the characterization of the molecular mechanisms comprising protein phosphorylation and cell signaling, culminating in neuroinflammation.
Keywords: Alzheimer’s disease; Parkinson’s disease; amyotrophic lateral sclerosis; astrocytes; cell signaling; gliosis; hydrocarbons; immune response; inflammation; metals; microglia; multiple sclerosis; nanoparticles; neurodegenerative diseases; neuroinflammation; neurological disorders; occupational injury; traumatic brain injury; workplace toxicants.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
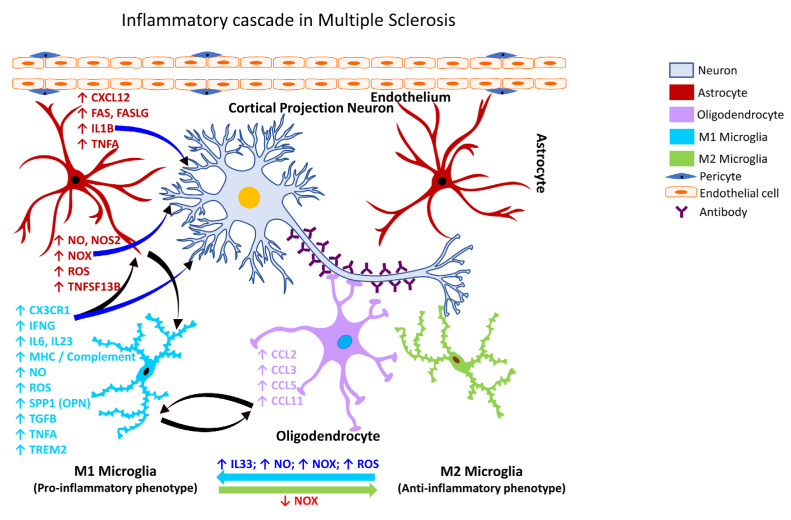
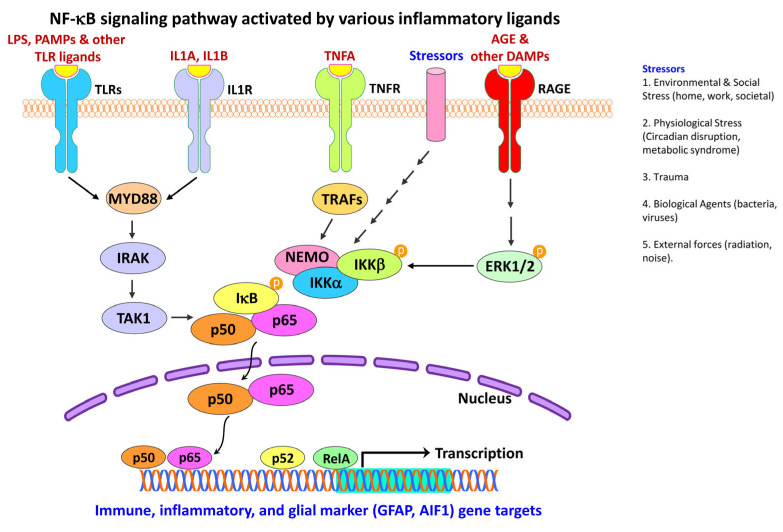
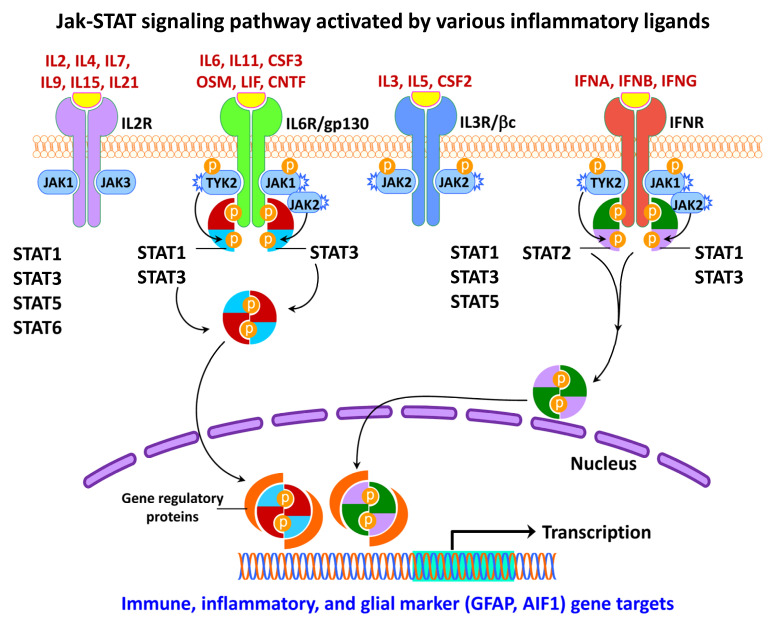
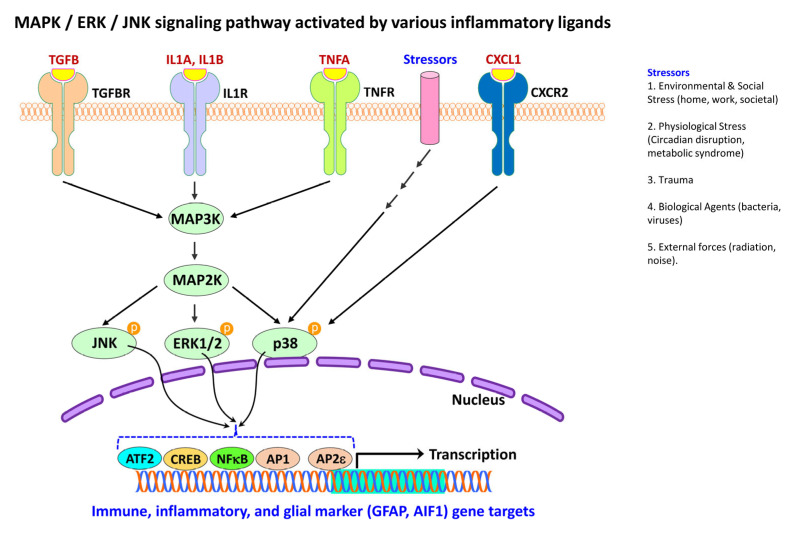
References
-
- Netea M.G., Balkwill F., Chonchol M., Cominelli F., Donath M.Y., Giamarellos-Bourboulis E.J., Golenbock D., Gresnigt M.S., Heneka M.T., Hoffman H.M., et al. A guiding map for inflammation. Nat. Immunol. 2017;18:826–831. doi: 10.1038/ni.3790. Erratum in Nat. Immunol. 2021, 22, 254. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
