

Serum KL-6 as a Biomarker of Progression at Any Time in Fibrotic Interstitial Lung Disease
- PMID: 36769819
- PMCID: PMC9917700
- DOI: 10.3390/jcm12031173
Serum KL-6 as a Biomarker of Progression at Any Time in Fibrotic Interstitial Lung Disease
Abstract
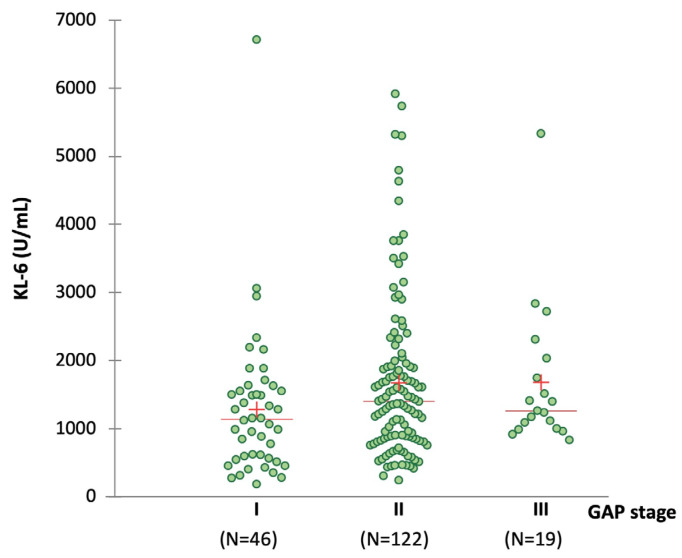
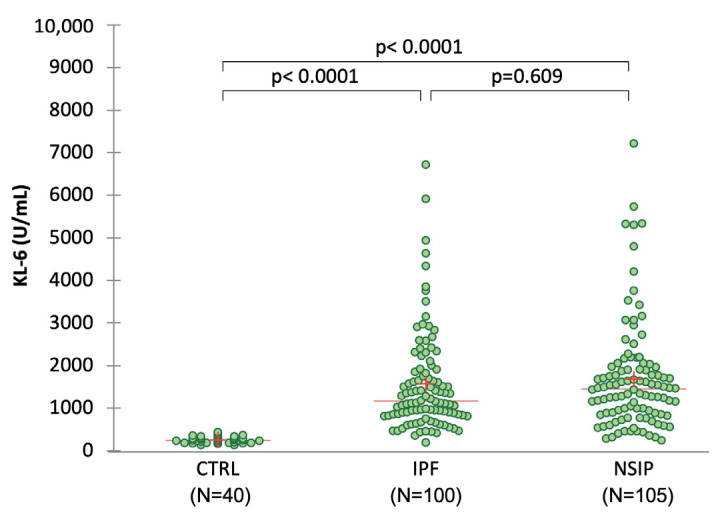
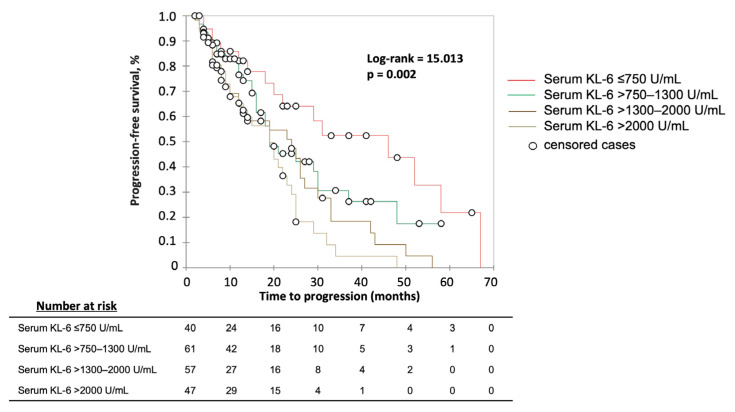
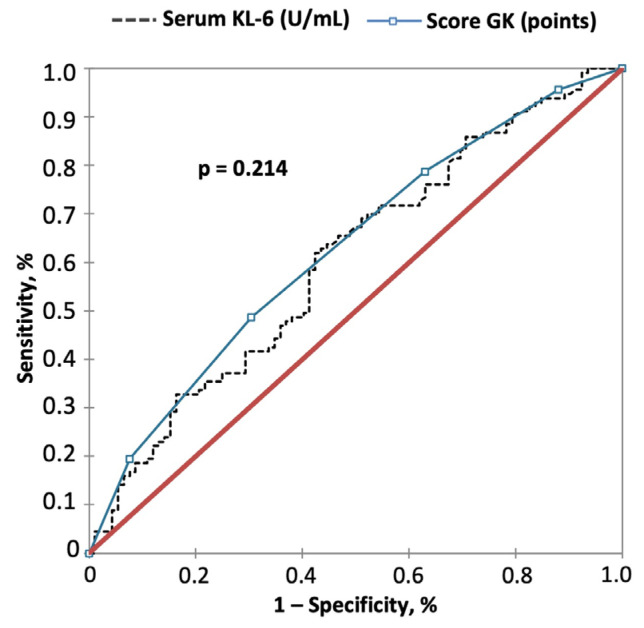
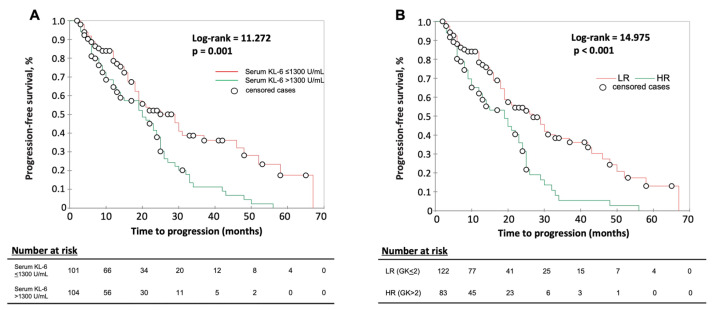
The development of a progressive phenotype of interstitial lung disease (ILD) is still unpredictable. Whereas tools to predict mortality in ILD exist, scores to predict disease progression are missing. The aim of this study was to investigate whether baseline serum KL-6 as an established marker to assess disease activity in ILD, alone or in combination with clinical variables, could improve stratification of ILD patients according to progression risk at any time. Consecutive patients with fibrotic ILD, followed at our institution between 2008 and 2015, were investigated. Disease progression was defined as relative decline of ≥10% in forced vital capacity (FVC) or ≥15% in diffusing capacity of the lung for carbon monoxide (DLco)% from baseline at any time. Serum KL-6 was measured using an automated immunoassay (Fujirebio Europe, Gent, Belgium). A stepwise logistic regression was performed to select variables to be included in the score. A total of 205 patients (49% idiopathic pulmonary fibrosis (IPF), 51% fibrotic nonspecific interstitial pneumonia (NSIP)) were included, of them 113 (55%) developed disease progression during follow up. Male gender (G) and serum KL-6 strata (K) were significant predictors of progression at regression analysis and were included in the GK score. A threshold of 2 GK score points was best for discriminating patients at high risk versus low risk to develop disease progression at any time. Serum KL-6 concentration, alone or combined in a simple score with gender, allows an effective stratification of ILD patients for risk of disease progression at any time.
Keywords: KL-6; biomarker; fibrotic ILD; progressive ILD.
Conflict of interest statement
L.B.J. reports travel costs reimbursement from Boehringer Ingelheim (BI) not related to the present work. U.C., C.T. and E.B. report no conflict of interest. J.W. reports travel costs reimbursement from BI and Novartis; speaker honoraria from MSD, BI and Roche, and advisory fees from Novartis not related to the present work. D.T. reports speaker honoraria from BI not related to the present work. F.B. reports speaker honoraria and advisory fees from Fujirebio Inc. related to the present work, speaker honoraria and travel costs reimbursement from BI and Roche, and advisory fees from BI, Roche, Bristol Myers Squibb, Galapagos, GlaxoSmithKline and Takeda not related to the present work.
Figures
References
-
- Brown K.K., Martinez F.J., Walsh S.L., Thannickal V.J., Prasse A., Schlenker-Herceg R., Goeldner R.-G., Clerisme-Beaty E., Tetzlaff K., Cottin V., et al. The natural history of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2020;55:2000085. doi: 10.1183/13993003.00085-2020. - DOI - PMC - PubMed
-
- Raghu G., Remy-Jardin M., Richeldi L., Thomson C.C., Inoue Y., Johkoh T., Kreuter M., Lynch D.A., Maher T.M., Martinez F.J., et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022;205:e18–e47. doi: 10.1164/rccm.202202-0399ST. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
