

Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies
- PMID: 36774340
- PMCID: PMC9922317
- DOI: 10.1038/s41419-023-05639-w
Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies
Abstract
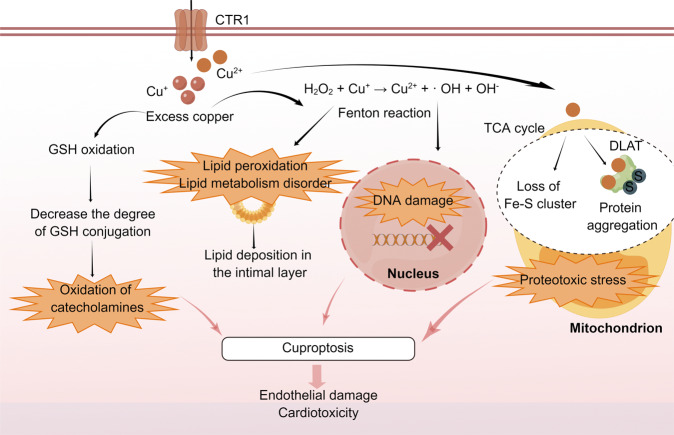
Copper is a vital mineral, and an optimal amount of copper is required to support normal physiologic processes in various systems, including the cardiovascular system. Over the past few decades, copper-induced cell death, named cuproptosis, has become increasingly recognized as an important process mediating the pathogenesis and progression of cardiovascular disease (CVD), including atherosclerosis, stroke, ischemia-reperfusion injury, and heart failure. Therefore, an in-depth understanding of the regulatory mechanisms of cuproptosis in CVD may be useful for improving CVD management. Here, we review the relationship between copper homeostasis and cuproptosis-related pathways in CVD, as well as therapeutic strategies addressing copper-induced cell death in CVD.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
