

The Amyloid Cascade Hypothesis 2.0: Generalization of the Concept
- PMID: 36777328
- PMCID: PMC9912825
- DOI: 10.3233/ADR-220079
The Amyloid Cascade Hypothesis 2.0: Generalization of the Concept
Abstract
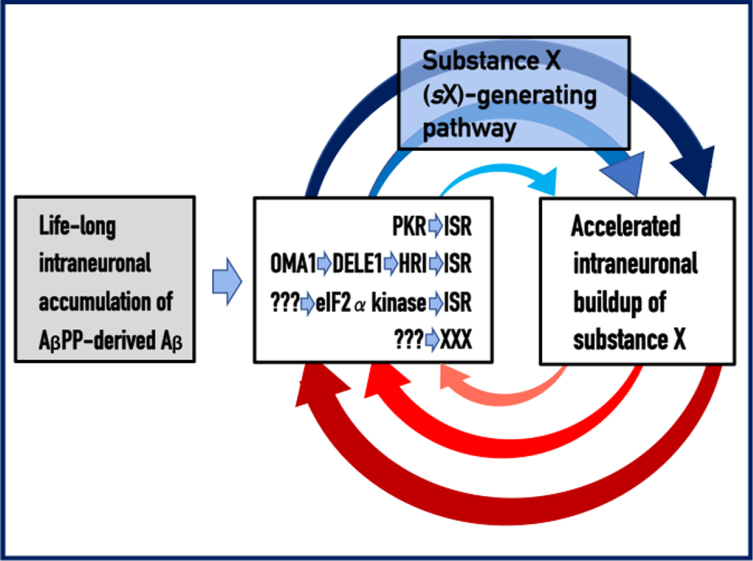
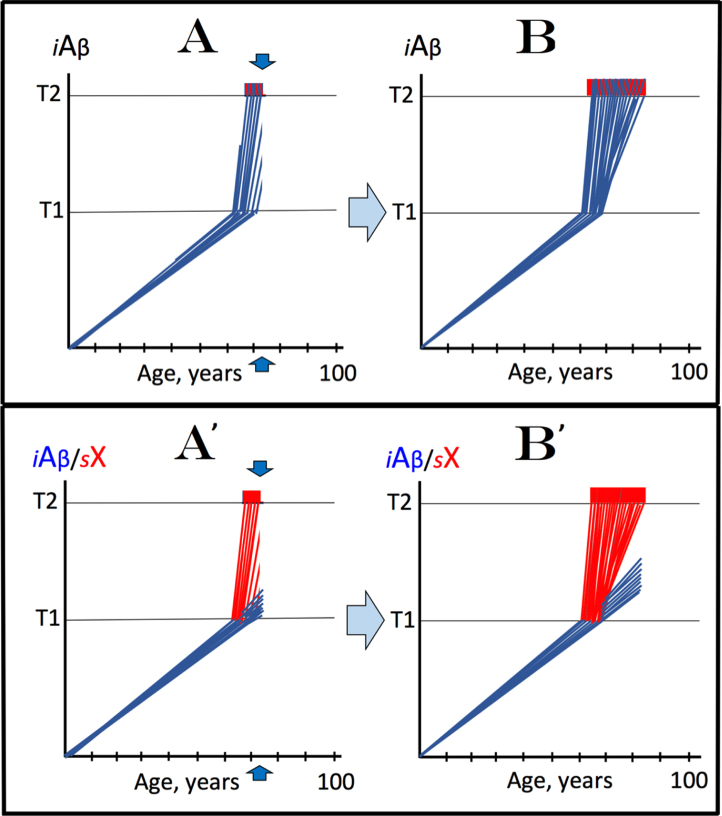
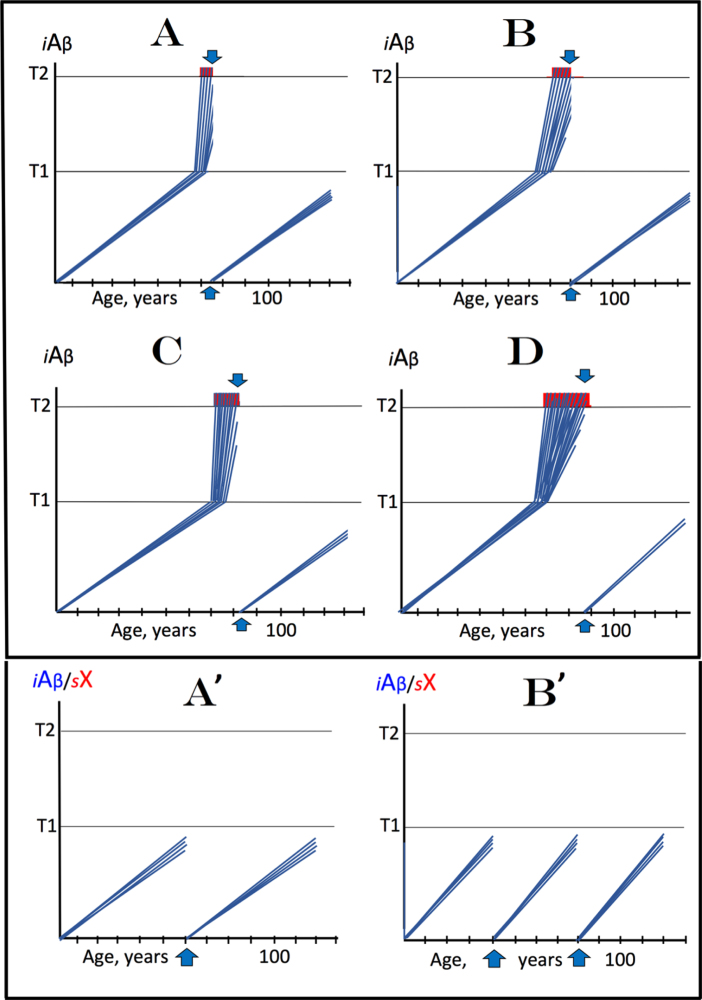
Recently, we proposed the Amyloid Cascade Hypothesis 2.0 (ACH2.0), a reformulation of the ACH. In the former, in contrast to the latter, Alzheimer's disease (AD) is driven by intraneuronal amyloid-β (iAβ) and occurs in two stages. In the first, relatively benign stage, Aβ protein precursor (AβPP)-derived iAβ activates, upon reaching a critical threshold, the AβPP-independent iAβ-generating pathway, triggering a devastating second stage resulting in neuronal death. While the ACH2.0 remains aligned with the ACH premise that Aβ is toxic, the toxicity is exerted because of intra- rather than extracellular Aβ. In this framework, a once-in-a-lifetime-only iAβ depletion treatment via transient activation of BACE1 and/or BACE2 (exploiting their Aβ-cleaving activities) or by any means appears to be the best therapeutic strategy for AD. Whereas the notion of differentially derived iAβ being the principal moving force at both AD stages is both plausible and elegant, a possibility remains that the second AD stage is enabled by an AβPP-derived iAβ-activated self-sustaining mechanism producing a yet undefined deleterious "substance X" (sX) which anchors the second AD stage. The present study generalizes the ACH2.0 by incorporating this possibility and shows that, in this scenario, the iAβ depletion therapy may be ineffective at symptomatic AD stages but fully retains its preventive potential for both AD and the aging-associated cognitive decline, which is defined in the ACH2.0 framework as the extended first stage of AD.
Keywords: Age-related cognitive decline; Alzheimer’s disease; Amyloid Cascade Hypothesis 2.0; BACE1 and BACE2 activators; amyloid-β protein precursor; intraneuronal amyloid-β.
© 2023 – The authors. Published by IOS Press.
Conflict of interest statement
The authors have no conflict of interest to report.
Figures
References
-
- Hardy JA, Higgins GA (1992) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185. - PubMed
-
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L (2004) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165, 1289–1300. - PMC - PubMed
-
- Bayer TA, Wirths O (2008) Review on the APP/PS1KI mouse model: Intraneuronal Abeta accumulation triggers opathy, neuron loss and working memory impairment. Genes Brain Behav 7, 6–11 axon. - PubMed
-
- Bayer TA, Breyhan H, Duan K, Rettig J, Wirths O (2008) Intraneuronal beta-amyloid is a major risk factor–novel evidence from the APP/PS1KI mouse model. Neurodegener Dis 5, 140–142. - PubMed
LinkOut - more resources
Full Text Sources
