

Clinical and genetic features of luscan-lumish syndrome associated with a novel de novo variant of SETD2 gene: Case report and literature review
- PMID: 36777730
- PMCID: PMC9911649
- DOI: 10.3389/fgene.2023.1081391
Clinical and genetic features of luscan-lumish syndrome associated with a novel de novo variant of SETD2 gene: Case report and literature review
Abstract
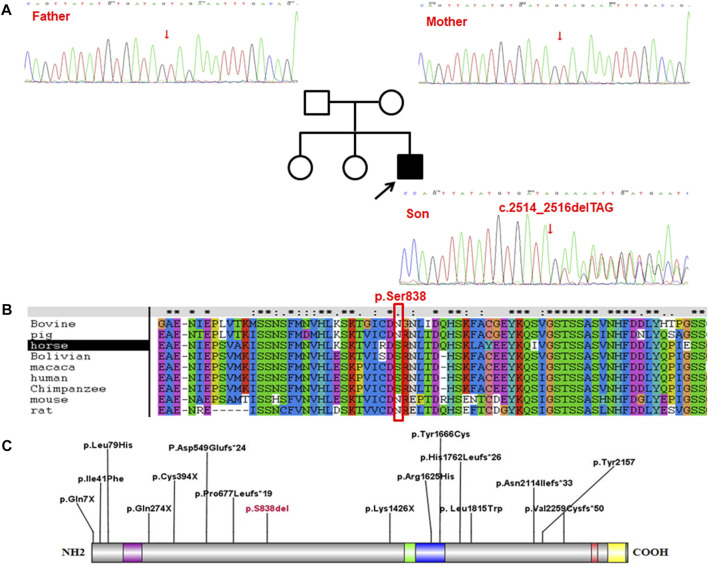
Introduction: Luscan-Lumish syndrome (LLS) is currently recognized as a rarely-observed condition featured with overgrowth, macrocephaly, obesity, type I Chiari malformation, and linguistic retardation. So far, there have been only a few LLS cases registered worldwide, but with none of them reported from China. To acquire a deeper understanding on the clinical and genetic features of this disease, a Chinese boy with LLS caused by a heterozygous variant in SETD2 gene was investigated in the present study. Methods: The patient was clinically examined and the medical history of his family was collected. Genetic testing was performed to determine the genetic etiology. Results: The proband was a boy aged 5-year-7-month-old, who was referred to our hospital due to "being a slow learner in kindergarten". The child had a history of delayed motor and language development in comparison to his peers. After admission, physical examination revealed tall stature and macrocephaly as the major manifestation, in addition to a relatively lower rating in intelligence assessment as well as abnormal MRI images showing a slightly shorter corpus callosum accompanied by a mildly thinner corpus callosum body. Whole exome sequencing (WES) revealed a heterozygous c.2514_2516delTAG (p.Ser838del) variant in SETD2 gene, which was subsequently identified as a novel de novo variant. According to the standardized genetic variant classification published by the American College of Medical Genetics and Genomics (ACMG), the variant, with a pathogenicity analysis result indicating PS2 + PM2_Supporting + PM4, was determined to be likely pathogenic. Through literature review, the clinical phenotypes of the 15 LLS cases were summarized, including 8 cases of overgrowth (53%), 13 cases of macrocephaly (87%), 11 cases of developmental delay (73%), 8 cases of autism (53%), and 7 cases of special facial features (47%). Besides, abnormal craniocerebral MRI findings were noticed in 7 cases. Despite that the mutation sites of the 15 patients varied from case to case, they showed a uniformly distributed pattern throughout the whole SETD2 gene, including 5 missense mutations, 5 frameshift mutations and 5 non-sense mutations. Conclusion: LLS, not having been recognized till recent years, is identified as an autosomal dominant syndrome triggered by SETD2 gene mutation. As the first report of LLS in China, the case in our study was proved to be associated with a unique type of SETD2 gene mutation that has never been reported previously, which is believed to enrich the mutation spectrum of SETD2 gene and also, deepening the clinicians' understanding on the disease.
Keywords: SETD2; developmental delay; gene mutation; luscan-lumish syndrome; macrocephaly; tall stature.
Copyright © 2023 Zhang, Zhang, Wu, Wang, Lv, Zhao, Wang, Liu and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
A novel SETD2 variant causing global development delay without overgrowth in a Chinese 3-year-old boy.Front Genet. 2023 Mar 21;14:1153284. doi: 10.3389/fgene.2023.1153284. eCollection 2023. Front Genet. 2023. PMID: 37025455 Free PMC article.
-
Clinical Heterogeneity and Different Phenotypes in Patients with SETD2 Variants: 18 New Patients and Review of the Literature.Genes (Basel). 2023 May 29;14(6):1179. doi: 10.3390/genes14061179. Genes (Basel). 2023. PMID: 37372360 Free PMC article. Review.
-
A Case of Luscan-Lumish Syndrome: Possible Involvement of Enhanced GH Signaling.J Clin Endocrinol Metab. 2021 Mar 8;106(3):718-723. doi: 10.1210/clinem/dgaa893. J Clin Endocrinol Metab. 2021. PMID: 33248444
-
Two novel cases expanding the phenotype of SETD2-related overgrowth syndrome.Am J Med Genet A. 2018 May;176(5):1212-1215. doi: 10.1002/ajmg.a.38666. Am J Med Genet A. 2018. PMID: 29681085
-
[Clinical analysis of a child with heterotopic ventricular gray matter Renpenning syndrome caused by PQBP1 gene mutation and a literature review].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025 Mar 10;42(3):314-321. doi: 10.3760/cma.j.cn511374-20240923-00502. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025. PMID: 40372223 Review. Chinese.
Cited by
-
Luscan-Lumish syndrome: A case report.World J Clin Cases. 2025 Jun 26;13(18):101471. doi: 10.12998/wjcc.v13.i18.101471. World J Clin Cases. 2025. PMID: 40574928 Free PMC article.
-
Disease spectrum, prevalence, genetic characteristics of inborn errors of metabolism in 21,840 hospitalized infants in Chongqing, China, 2017-2022.Front Genet. 2024 May 28;15:1395988. doi: 10.3389/fgene.2024.1395988. eCollection 2024. Front Genet. 2024. PMID: 38863445 Free PMC article.
-
Mechanisms of brain overgrowth in autism spectrum disorder with macrocephaly.Front Neurosci. 2025 Jun 6;19:1586550. doi: 10.3389/fnins.2025.1586550. eCollection 2025. Front Neurosci. 2025. PMID: 40548072 Free PMC article. Review.
References
-
- Hacker K. E., Fahey C. C., Shinsky S. A., Chiang Y. J., DiFiore J. V., Jha D. K., et al. (2016). Structure/function analysis of recurrent mutations in SETD2 protein reveals a critical and conserved role for a SET domain residue in maintaining protein stability and histone H3 lys-36 trimethylation. J. Biol. Chem. 291, 21283–21295. 10.1074/jbc.M116.739375 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials