

Expanding the genomic encyclopedia of Actinobacteria with 824 isolate reference genomes
- PMID: 36778052
- PMCID: PMC9903846
- DOI: 10.1016/j.xgen.2022.100213
Expanding the genomic encyclopedia of Actinobacteria with 824 isolate reference genomes
Abstract
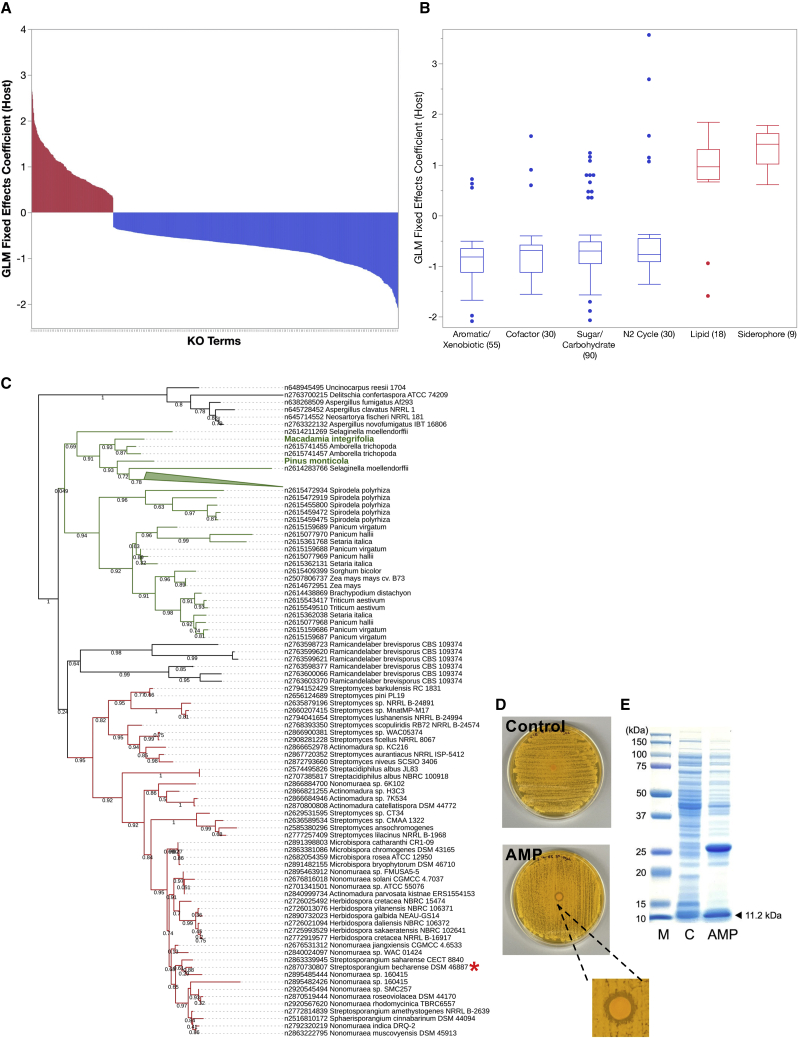
The phylum Actinobacteria includes important human pathogens like Mycobacterium tuberculosis and Corynebacterium diphtheriae and renowned producers of secondary metabolites of commercial interest, yet only a small part of its diversity is represented by sequenced genomes. Here, we present 824 actinobacterial isolate genomes in the context of a phylum-wide analysis of 6,700 genomes including public isolates and metagenome-assembled genomes (MAGs). We estimate that only 30%-50% of projected actinobacterial phylogenetic diversity possesses genomic representation via isolates and MAGs. A comparison of gene functions reveals novel determinants of host-microbe interaction as well as environment-specific adaptations such as potential antimicrobial peptides. We identify plasmids and prophages across isolates and uncover extensive prophage diversity structured mainly by host taxonomy. Analysis of >80,000 biosynthetic gene clusters reveals that horizontal gene transfer and gene loss shape secondary metabolite repertoire across taxa. Our observations illustrate the essential role of and need for high-quality isolate genome sequences.
Keywords: actinobacteria; comparative genomics; ecology; evolution; metagenomics; microbiology; mycobacteria; secondary metabolites.
© 2022 The Authors.
Conflict of interest statement
The authors declare no competing financial interests.
Figures





References
-
- Navarro-Muñoz J.C., Selem-Mojica N., Mullowney M.W., Kautsar S.A., Tryon J.H., Parkinson E.I., De Los Santos E.L.C., Yeong M., Cruz-Morales P., Abubucker S., et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020;16:60–68. doi: 10.1038/s41589-019-0400-9. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
