

This is a preprint.
Large scale genome-wide association analyses identify novel genetic loci and mechanisms in hypertrophic cardiomyopathy
- PMID: 36778260
- PMCID: PMC9915807
- DOI: 10.1101/2023.01.28.23285147
Large scale genome-wide association analyses identify novel genetic loci and mechanisms in hypertrophic cardiomyopathy
Update in
-
Large-scale genome-wide association analyses identify novel genetic loci and mechanisms in hypertrophic cardiomyopathy.Nat Genet. 2025 Mar;57(3):530-538. doi: 10.1038/s41588-025-02087-4. Epub 2025 Feb 18. Nat Genet. 2025. PMID: 39966646 Free PMC article.
Abstract
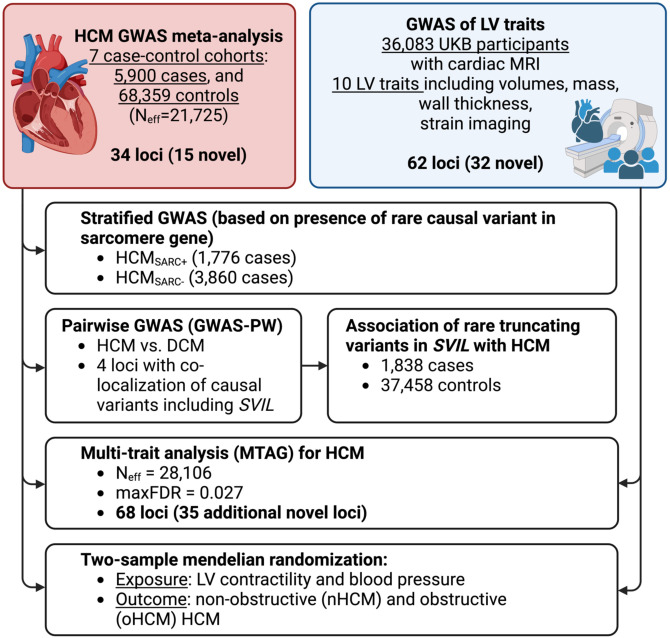
Hypertrophic cardiomyopathy (HCM) is an important cause of morbidity and mortality with both monogenic and polygenic components. We here report results from the largest HCM genome-wide association study (GWAS) and multi-trait analysis (MTAG) including 5,900 HCM cases, 68,359 controls, and 36,083 UK Biobank (UKB) participants with cardiac magnetic resonance (CMR) imaging. We identified a total of 70 loci (50 novel) associated with HCM, and 62 loci (32 novel) associated with relevant left ventricular (LV) structural or functional traits. Amongst the common variant HCM loci, we identify a novel HCM disease gene, SVIL, which encodes the actin-binding protein supervillin, showing that rare truncating SVIL variants cause HCM. Mendelian randomization analyses support a causal role of increased LV contractility in both obstructive and non-obstructive forms of HCM, suggesting common disease mechanisms and anticipating shared response to therapy. Taken together, the findings significantly increase our understanding of the genetic basis and molecular mechanisms of HCM, with potential implications for disease management.
Figures




References
Publication types
Grants and funding
- CH/1992001/6764/BHF_/British Heart Foundation/United Kingdom
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- BBC/F/21/220106/BHF_/British Heart Foundation/United Kingdom
- FS/IPBSRF/22/27059/BHF_/British Heart Foundation/United Kingdom
- U01 HL117006/HL/NHLBI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
- SP/19/2/34462/BHF_/British Heart Foundation/United Kingdom
- NH/17/1/32725/BHF_/British Heart Foundation/United Kingdom
- MC_U120085815/MRC_/Medical Research Council/United Kingdom
- FS/ICRF/21/26019/BHF_/British Heart Foundation/United Kingdom
- MC_UP_1605/13/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous