

This is a preprint.
CDK4/6 inhibition enhances SHP2 inhibitor efficacy and is dependent upon restoration of RB function in malignant peripheral nerve sheath tumors
- PMID: 36778419
- PMCID: PMC9915673
- DOI: 10.1101/2023.02.02.526674
CDK4/6 inhibition enhances SHP2 inhibitor efficacy and is dependent upon restoration of RB function in malignant peripheral nerve sheath tumors
Update in
-
CDK4/6 inhibition enhances SHP2 inhibitor efficacy and is dependent upon RB function in malignant peripheral nerve sheath tumors.Sci Adv. 2023 Nov 24;9(47):eadg8876. doi: 10.1126/sciadv.adg8876. Epub 2023 Nov 24. Sci Adv. 2023. PMID: 38000020 Free PMC article.
Abstract
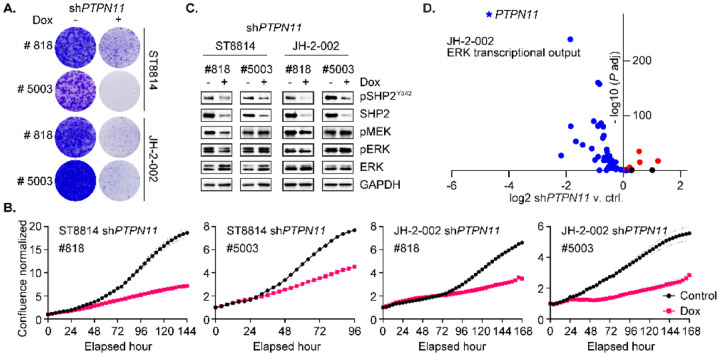
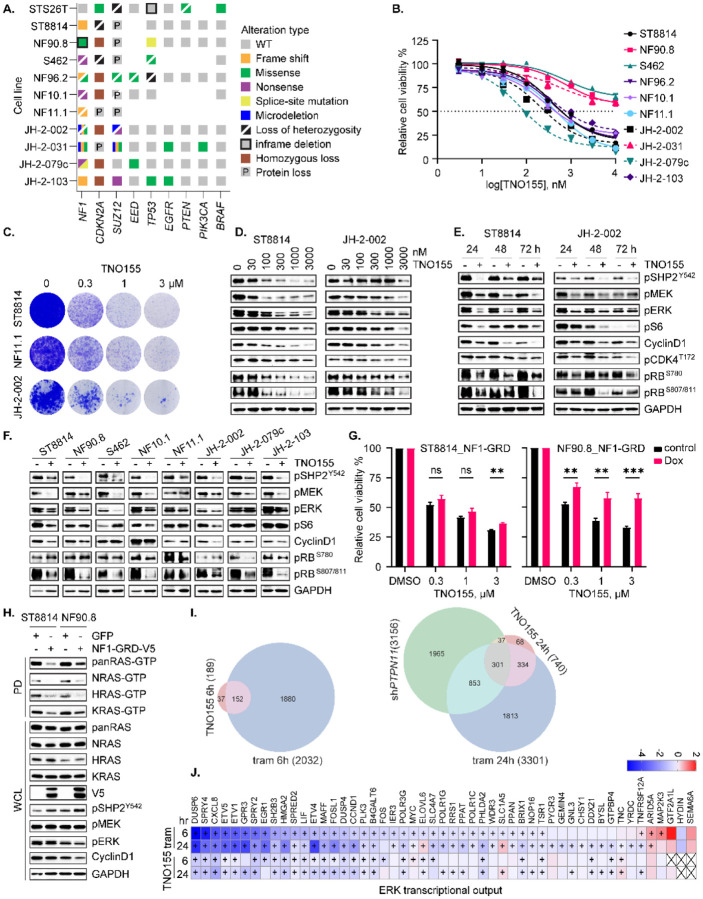
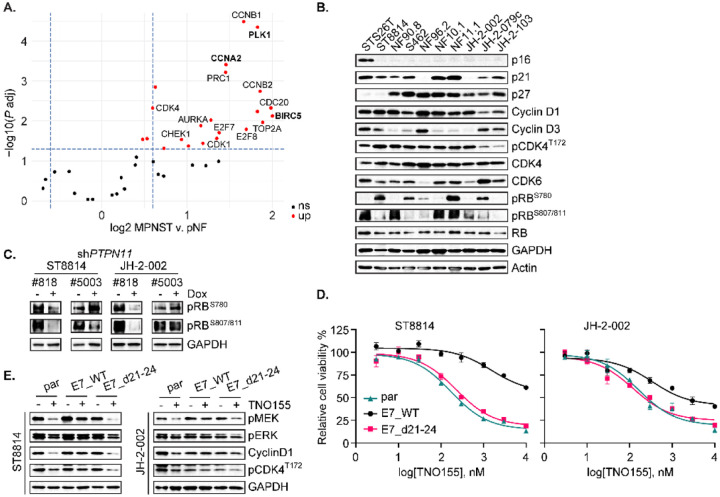
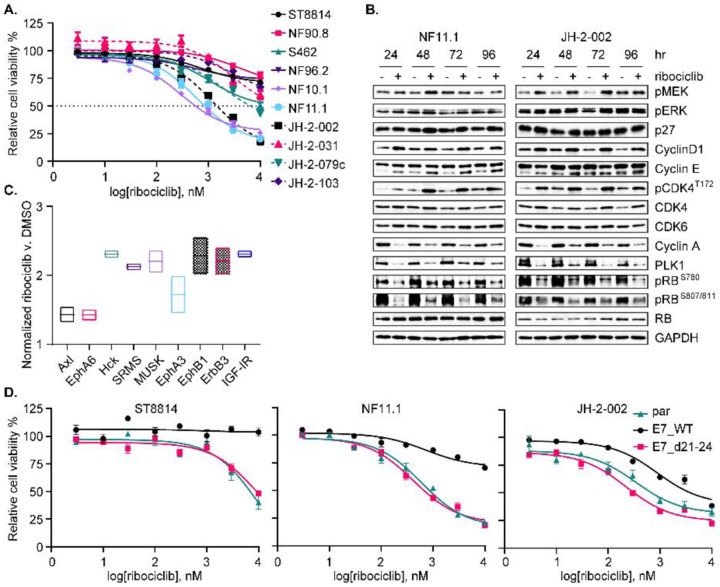
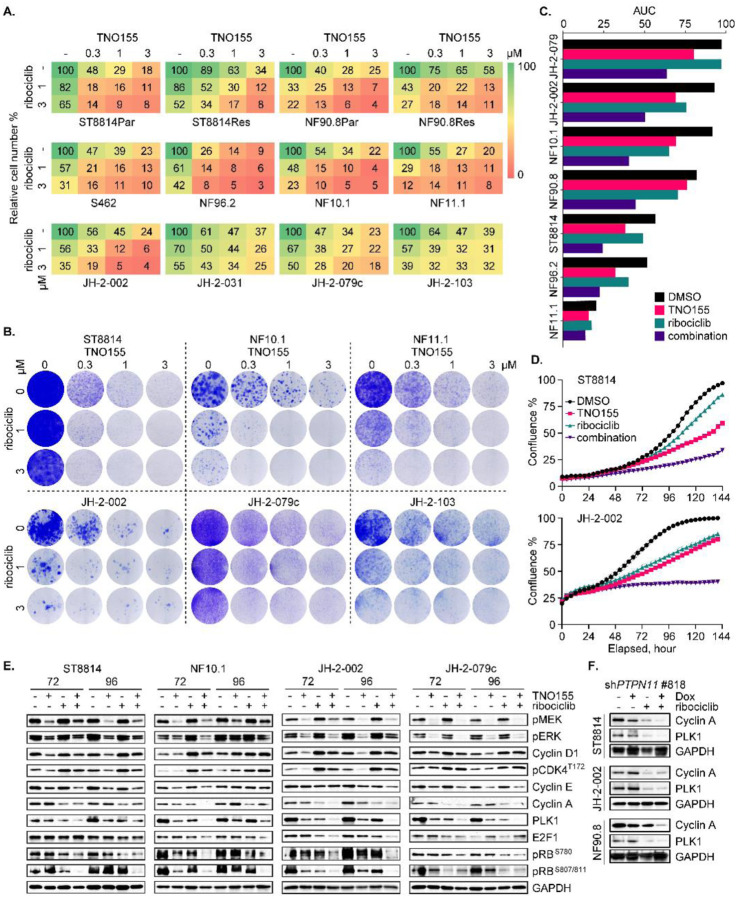
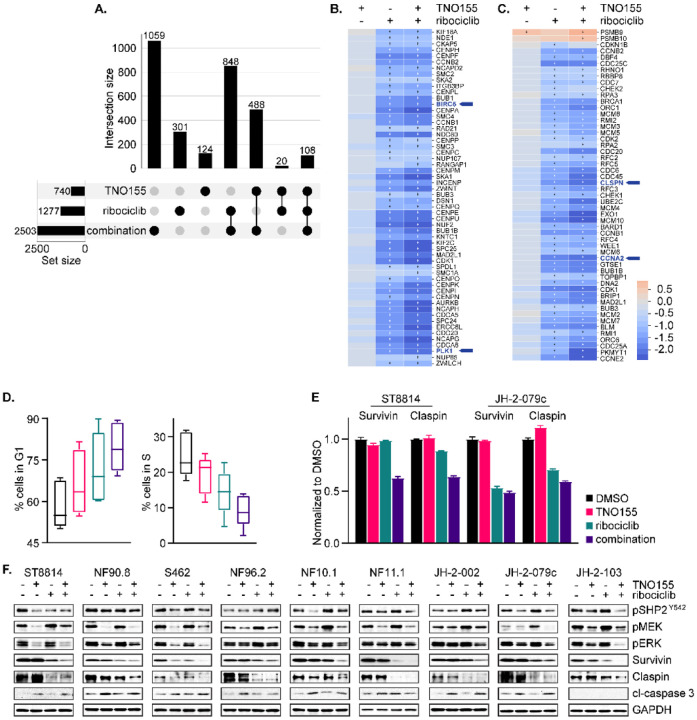
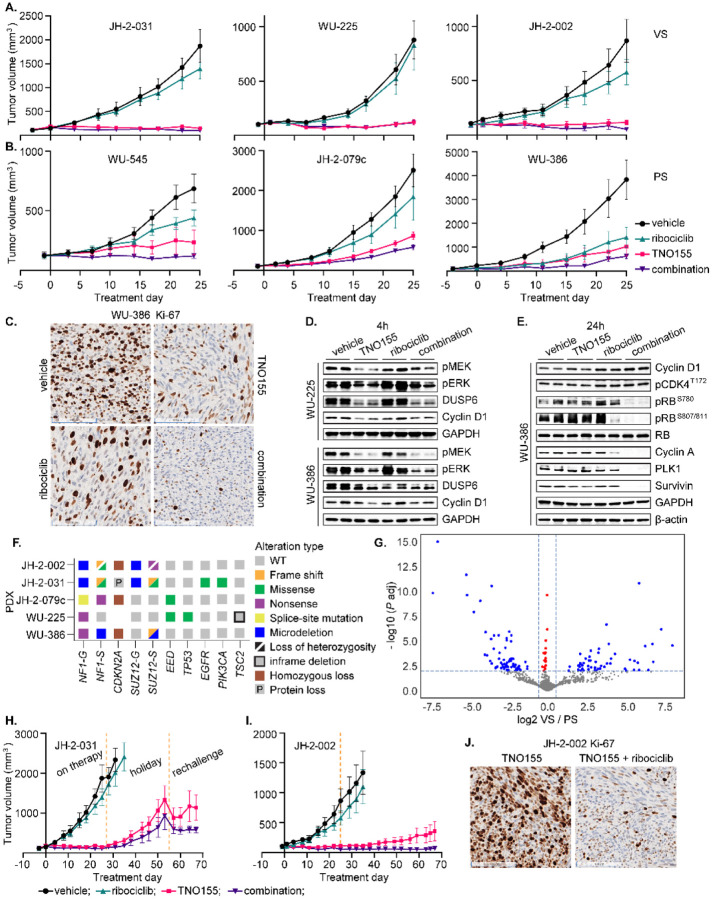
Malignant peripheral nerve sheath tumors (MPNST) are highly aggressive soft tissue sarcomas with limited treatment options, and novel effective therapeutic strategies are desperately needed. We observe anti-proliferative efficacy of genetic depletion or pharmacological inhibition using the clinically available SHP2 inhibitor (SHP2i) TNO155. Our studies into the signaling response to SHP2i reveal that resistance to TNO155 is partially mediated by reduced RB function, and we therefore test the addition of a CDK4/6 inhibitor (CDK4/6i) to enhance RB activity and improve TNO155 efficacy. In combination, TNO155 attenuates the adaptive response to CDK4/6i, potentiates its anti-proliferative effects, and converges on enhancement of RB activity, with greater suppression of cell cycle and inhibitor-of-apoptosis proteins, leading to deeper and more durable anti-tumor activity in in vitro and in vivo patient-derived models of MPNST, relative to either single agent. Overall, our study provides timely evidence to support the clinical advancement of this combination strategy in patients with MPNST and other tumors driven by loss of NF1.
Conflict of interest statement
Competing interests: JW, SEM and CAP have a pending patent related to this study. The authors have no additional financial interests.
Figures
References
-
- Ducatman B. S., Scheithauer B. W., Piepgras D. G., Reiman H. M., Ilstrup D. M., Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 57, 2006–2021 (1986). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous