

Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes
- PMID: 36778668
- PMCID: PMC9903662
- DOI: 10.1016/j.xgen.2022.100168
Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes
Abstract
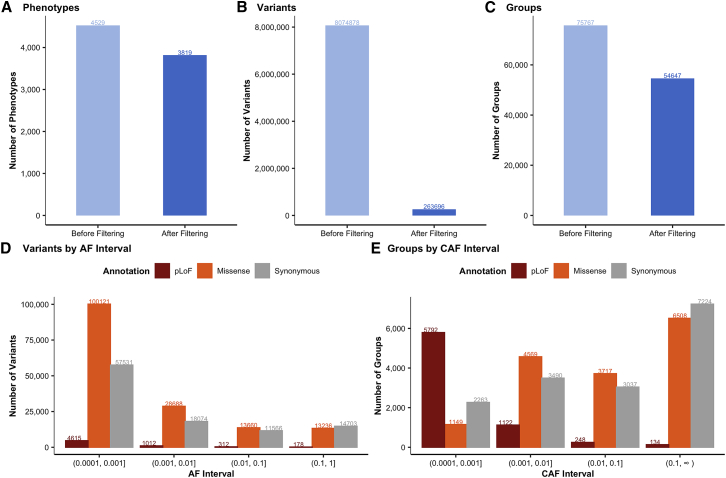
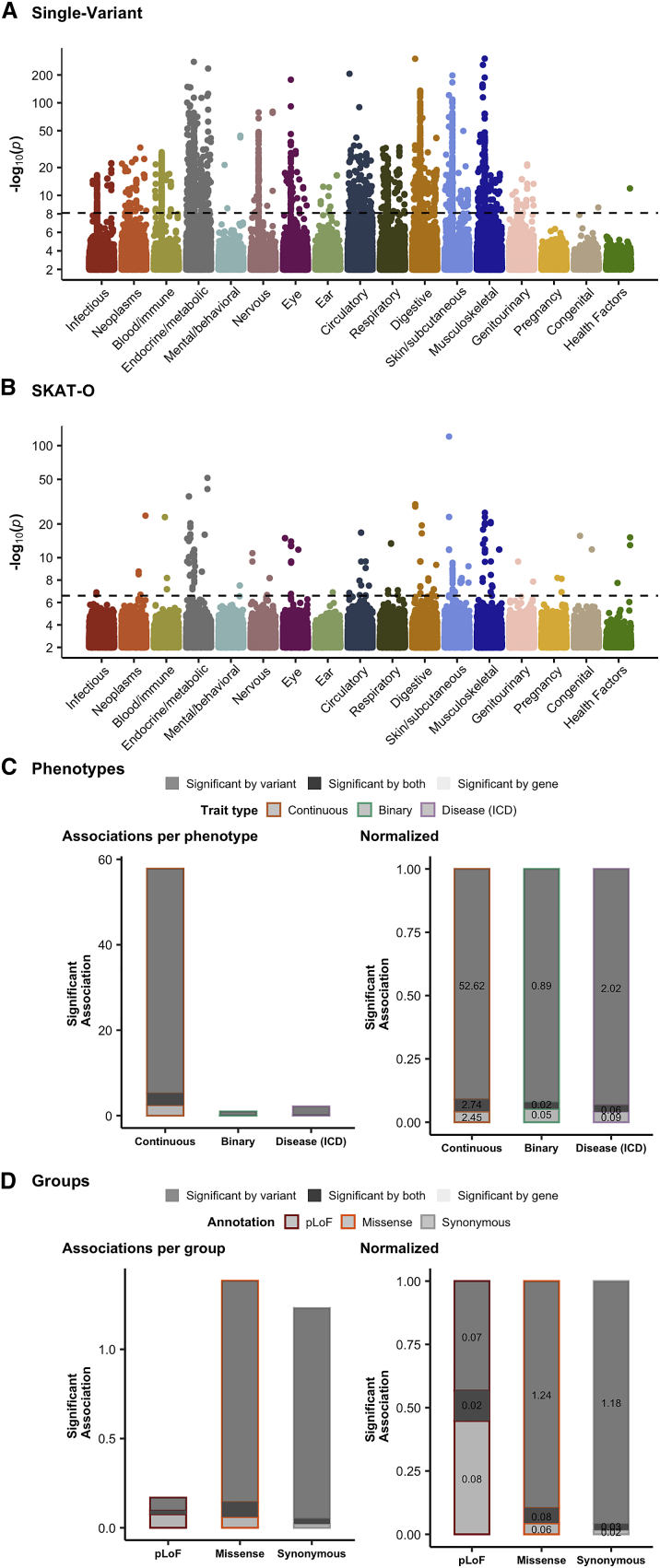
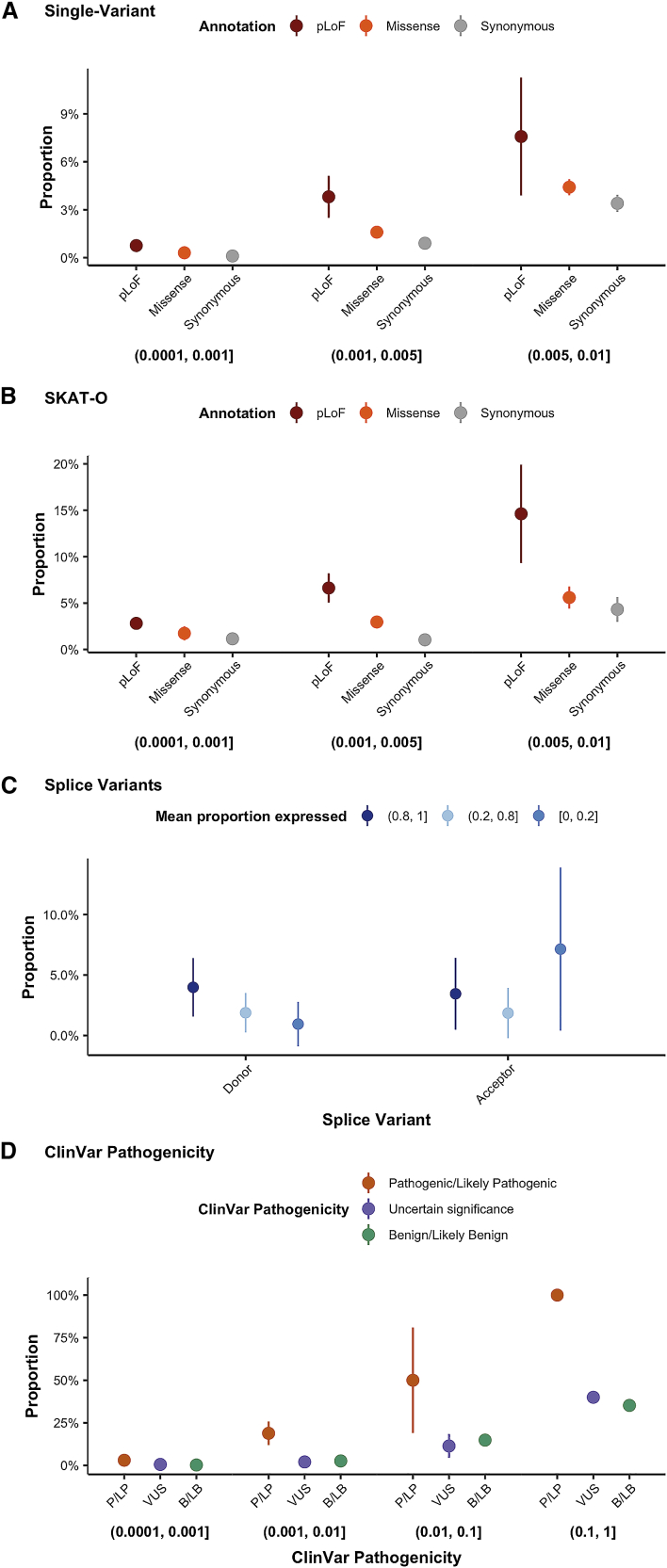
Genome-wide association studies have successfully discovered thousands of common variants associated with human diseases and traits, but the landscape of rare variations in human disease has not been explored at scale. Exome-sequencing studies of population biobanks provide an opportunity to systematically evaluate the impact of rare coding variations across a wide range of phenotypes to discover genes and allelic series relevant to human health and disease. Here, we present results from systematic association analyses of 4,529 phenotypes using single-variant and gene tests of 394,841 individuals in the UK Biobank with exome-sequence data. We find that the discovery of genetic associations is tightly linked to frequency and is correlated with metrics of deleteriousness and natural selection. We highlight biological findings elucidated by these data and release the dataset as a public resource alongside the Genebass browser for rapidly exploring rare-variant association results.
Keywords: GWAS; PheWAS; biobanks; exome sequencing; rare variant association studies; rare variants.
© 2022 The Author(s).
Conflict of interest statement
K.J.K. is a consultant for Vor Biopharma. B.M.R.-G., X.Z., F.R., S.E., A.J.G., M.R., J.W., H.J., and J.W.D. are employees of AbbVie, Inc. M.R. is an employee of and owns stock in AbbVie, Inc. E.A.T., D.S., P.G.B., and H.R. are employees of Biogen and hold stocks/stock options in Biogen. H.I.K., X.C., X.H., and M.R.M. are employees of Pfizer. D.K. holds stock in the private company TriNetX, LLC. D.S.P. was an employee of Genomics plc. All the analyses reported in this paper were performed as part of D.S.P.’s previous employment at the Massachusetts General Hospital and Broad Institute. N.A.W. owns stock in Pfizer. L.D.G. receives funding from Intel and Illumina. D.G.M. is a founder with equity of Goldfinch Bio and serves as a paid advisor to GSK, Variant Bio, Insitro, and Foresite Labs. H.L.R. is a member of the scientific advisory board at Genome Medical. A.A.P. is a Venture Partner at GV. He has received consulting fees from Novartis and receives funding from Bayer, IBM, Microsoft, Alphabet, Intel, GSK, Pfizer, and Illumina. M.J.D. is a founder of Maze Therapeutics. B.M.N. is a member of the scientific advisory board at Deep Genomics and RBNC Therapeutics, a member of the scientific advisory committee at Milken, and a consultant for Camp4 Therapeutics, Merck, and Biogen.
Figures
Comment in
-
Rare-variant collapsing analyses of arterial hypertension in the UK biobank.J Hum Hypertens. 2023 Nov;37(11):1040-1042. doi: 10.1038/s41371-023-00829-7. Epub 2023 Apr 14. J Hum Hypertens. 2023. PMID: 37059828 Free PMC article. No abstract available.
References
-
- Kalia S.S., Adelman K., Bale S.J., Chung W.K., Eng C., Evans J.P., Herman G.E., Hufnagel S.B., Klein T.E., Korf B.R., et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0. Genet. Med. 2017;19:249–255. - PubMed
-
- Abifadel M., Varret M., Rabès J.P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003;34:154–156. - PubMed
-
- Cohen J.C., Boerwinkle E., Mosley T.H., Jr., Hobbs H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. - PubMed
-
- Sabatine M.S., Giugliano R.P., Keech A.C., Honarpour N., Wiviott S.D., Murphy S.A., Kuder J.F., Wang H., Liu T., Wasserman S.M., et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. N. Engl. J. Med. 2017;376:1713–1722. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
