

Dynamic changes in fecal microbiota in donkey foals during weaning: From pre-weaning to post-weaning
- PMID: 36778861
- PMCID: PMC9915154
- DOI: 10.3389/fmicb.2023.1105330
Dynamic changes in fecal microbiota in donkey foals during weaning: From pre-weaning to post-weaning
Abstract
Introduction: A better understanding of the microbiota community in donkey foals during the weaning transition is a prerequisite to optimize gut function and improve feed efficiency. The objective of the present study was to investigate the dynamic changes in fecal microbiota in donkey foals from pre-to post-weaning period.
Methods: A total of 27 fecal samples of donkey foals were collected in the rectum before morning feeding at pre-weaning (30 days of age, PreW group, n = 9), dur-weaning (100 days of age, DurW group, n = 9) and post-weaning (170 days of age, PostW group, n = 9) period. The 16S rRNA amplicon sequencing were employed to indicate the microbial changes during the weaning period.
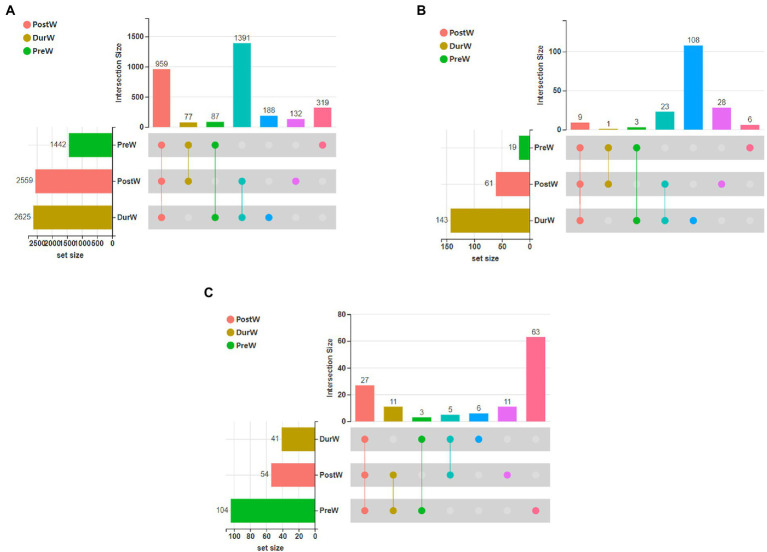
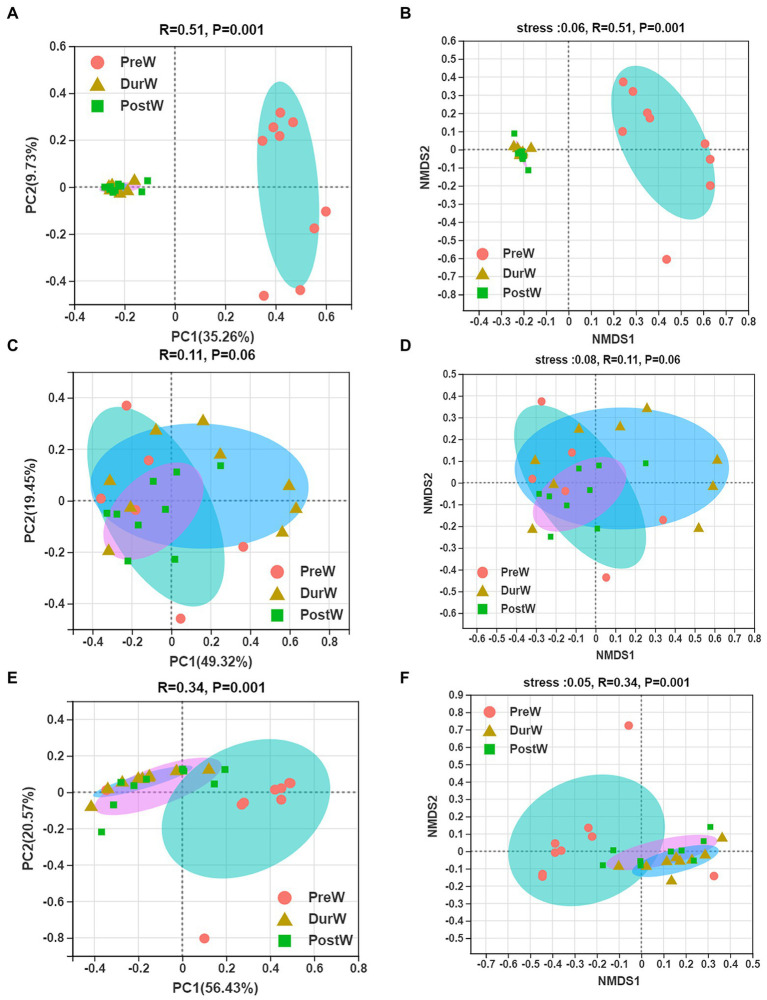
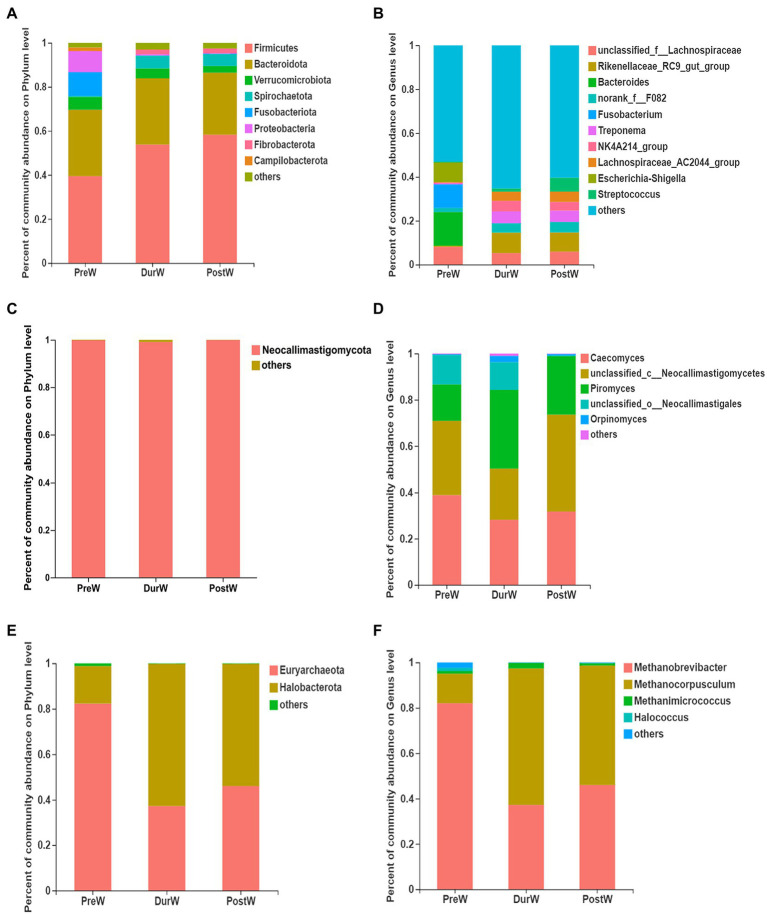
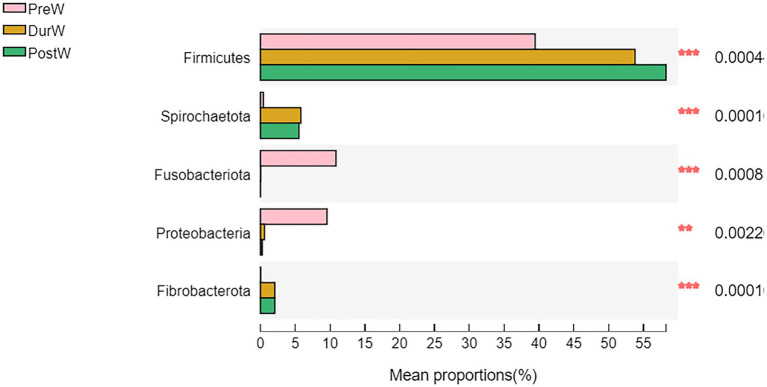
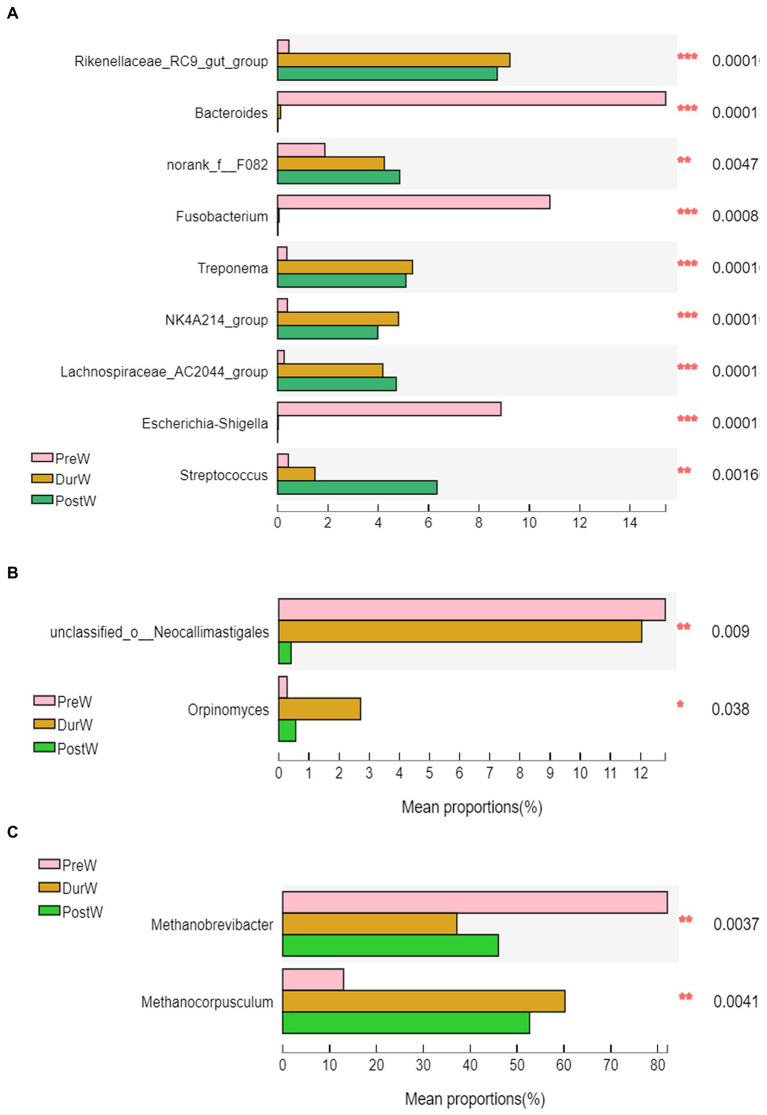
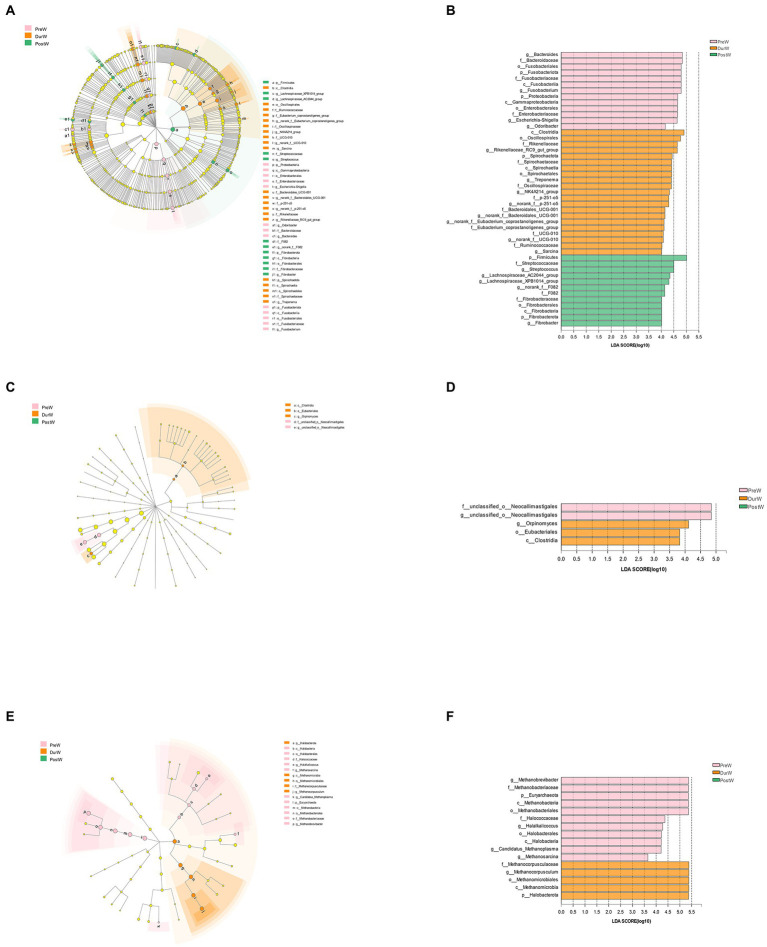
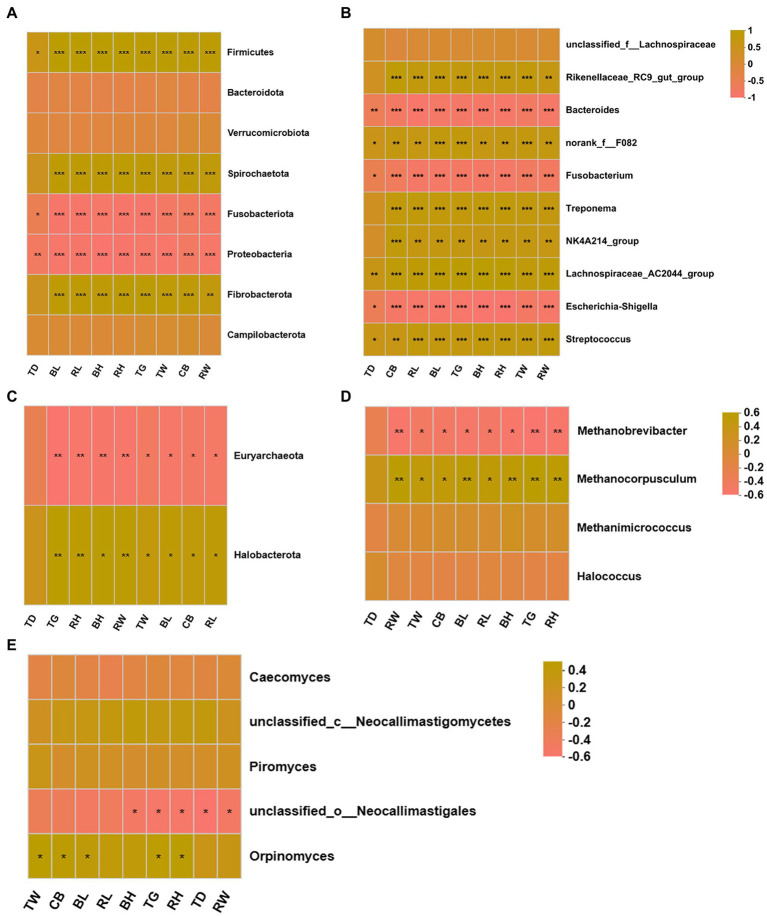
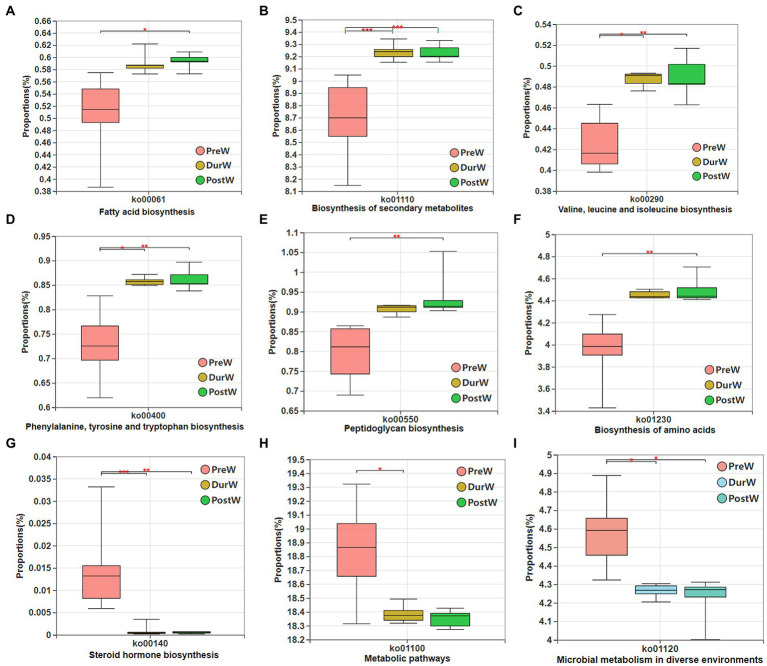
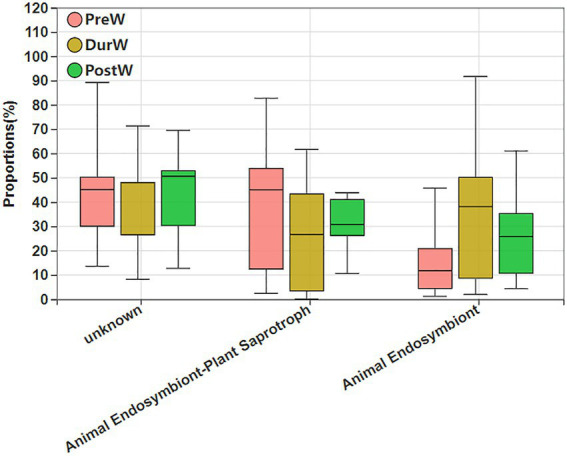
Results: In the present study, the cessation of breastfeeding gradually and weaning onto plant-based feeds increased the microbial diversity and richness, with a higher Shannon, Ace, Chao and Sobs index in DurW and PostW than in PreW (p < 0.05). The predominant bacterial phyla in donkey foal feces were Firmicutes (>50.5%) and Bacteroidota (>29.5%), and the predominant anaerobic fungi and archaea were Neocallimastigomycota and Euryarchaeota. The cellulolytic related bacteria including phylum Firmicutes, Spirochaetota and Fibrobacterota and genus norank_f_F082, Treponema, NK4A214_group, Lachnospiraceae_AC2044_group and Streptococcus were increased from pre-to post-weaning donkey foals (p < 0.05). Meanwhile, the functions related to the fatty acid biosynthesis, carbohydrate metabolism and amino acid biosynthesis were significantly enriched in the fecal microbiome in the DurW and PostW donkeys. Furthermore, the present study provided the first direct evidence that the initial colonization and establishment of anaerobic fungi and archaea in donkey foals began prior to weaning. The relative abundance of Orpinomyces were the highest in DurW donkey foals among the three groups (p < 0.01). In terms of archaea, the abundance of Methanobrevibacter were higher in PreW than in DurW and PostW (p < 0.01), but the abundance of Methanocorpusculum were significantly increased in DurW and PostW compared to PreW donkey foals (p < 0.01).
Discussion: Altogether, the current study contributes to a comprehensive understanding of the development of the microbiota community in donkey foals from pre-to post-weaning period, which may eventually result in an improvement of the digestion and feed efficiency in donkeys.
Keywords: anaerobic fungi; archaea; bacteria; donkey foal; fecal microbiota; weaning.
Copyright © 2023 Zhang, Huang, Gao, Shi, Wang, Wang, Wang, Liu and Wang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous