

Statins inhibit protein kinase D (PKD) activation in intestinal cells and prevent PKD1-induced growth of murine enteroids
- PMID: 36779664
- PMCID: PMC10042602
- DOI: 10.1152/ajpcell.00286.2022
Statins inhibit protein kinase D (PKD) activation in intestinal cells and prevent PKD1-induced growth of murine enteroids
Abstract
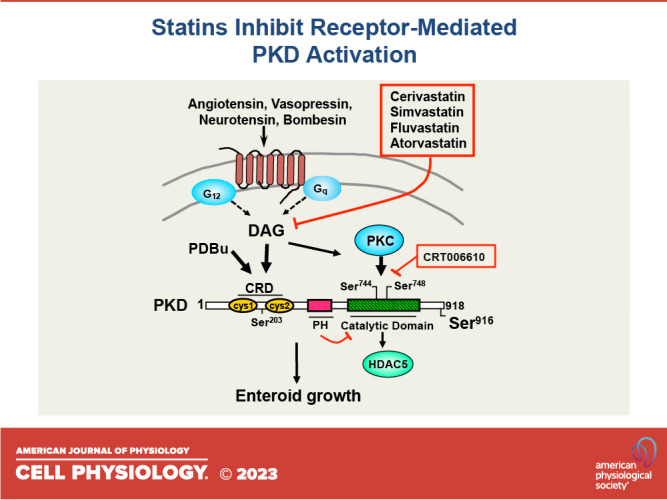
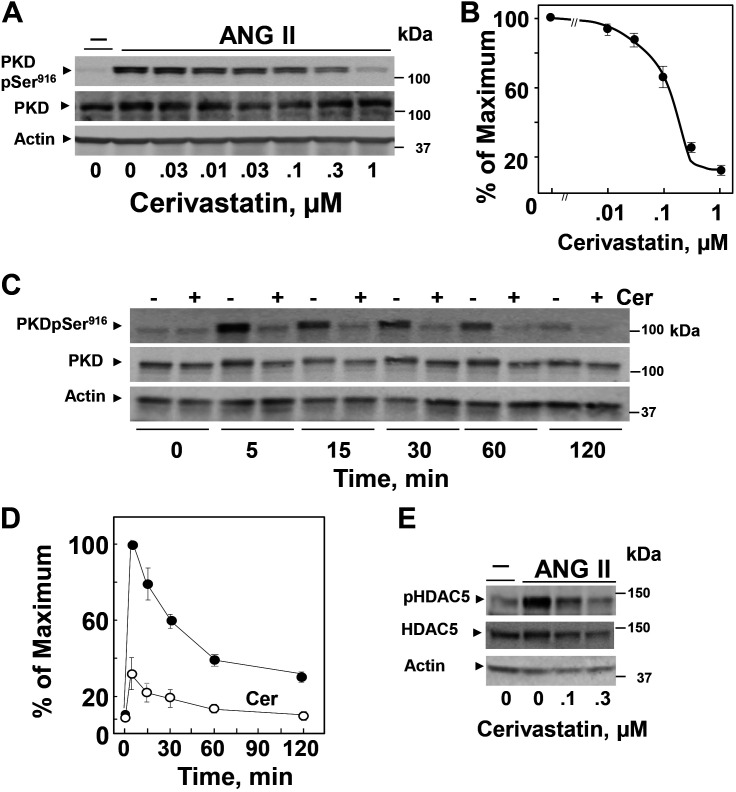
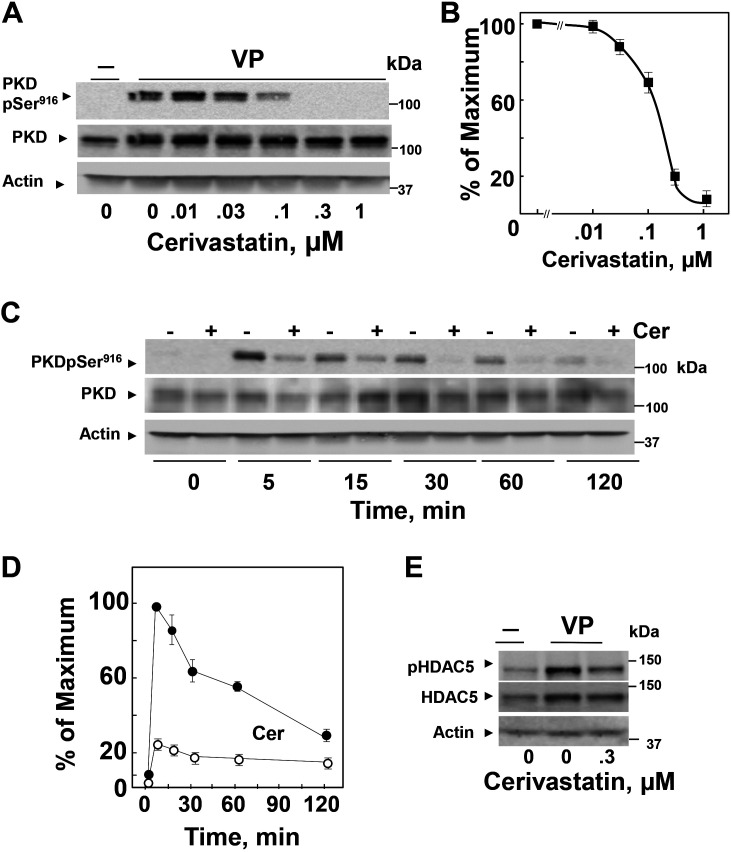
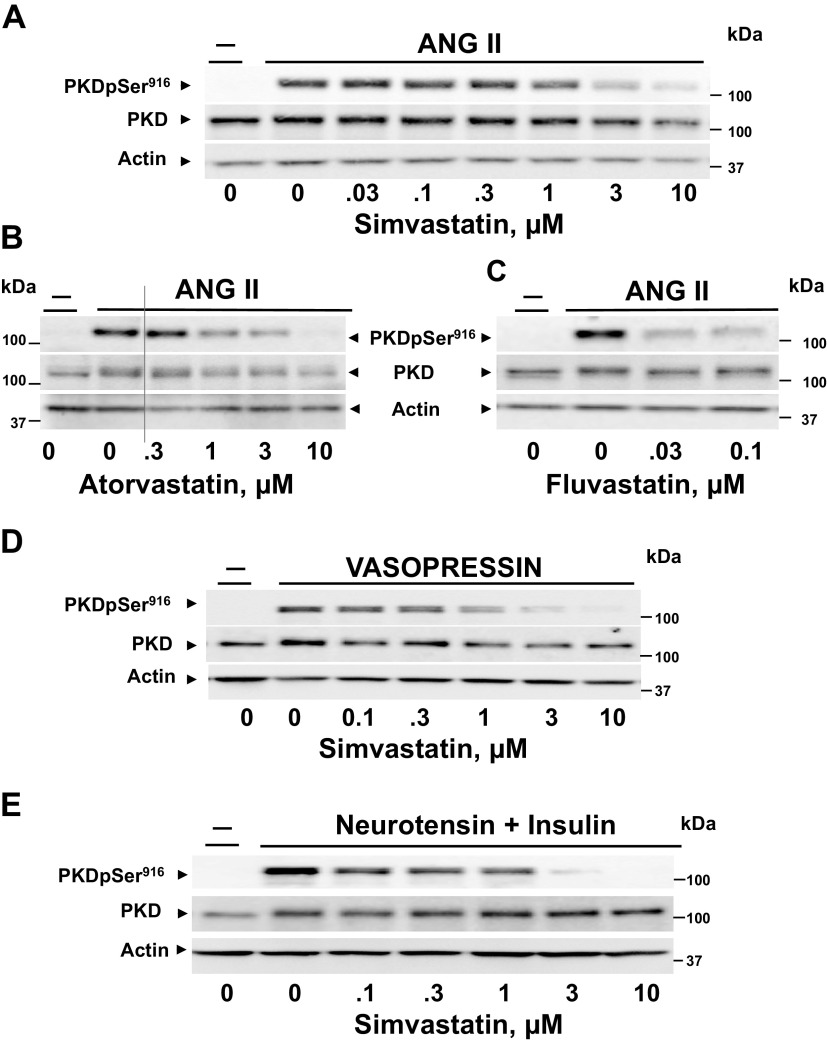
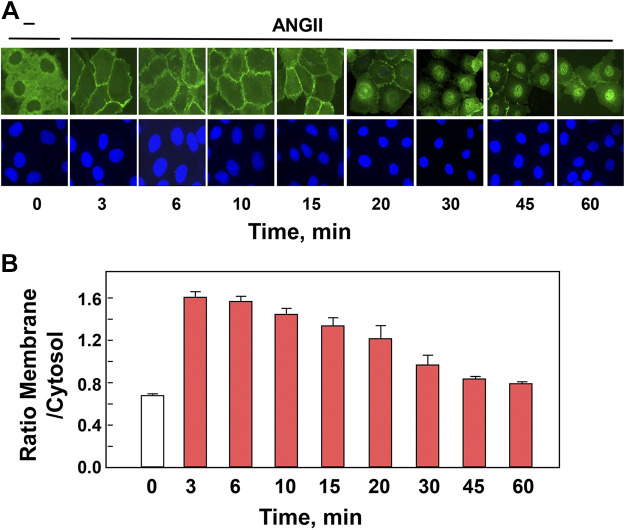
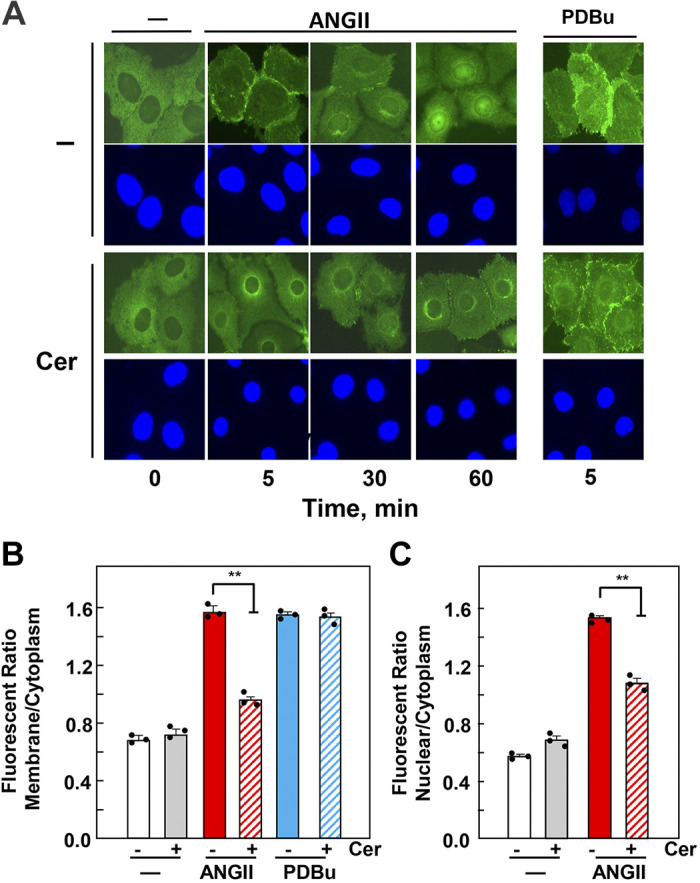
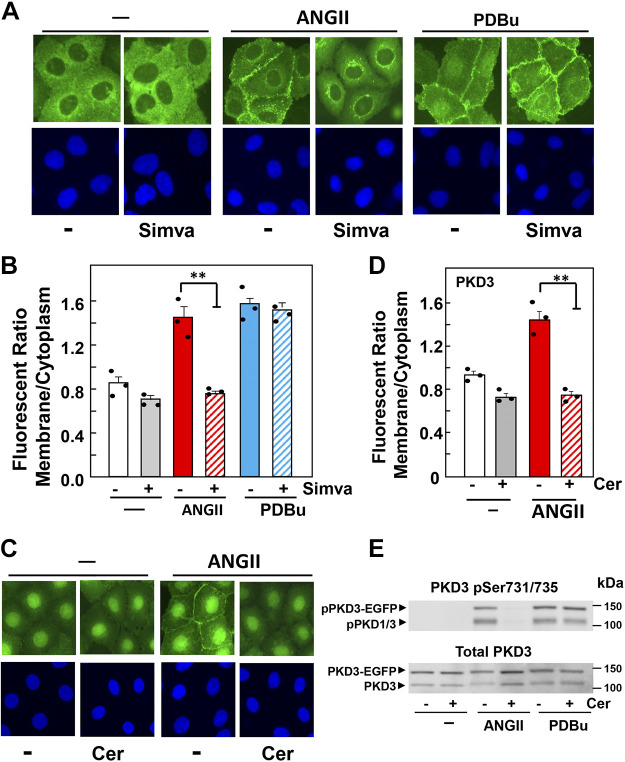
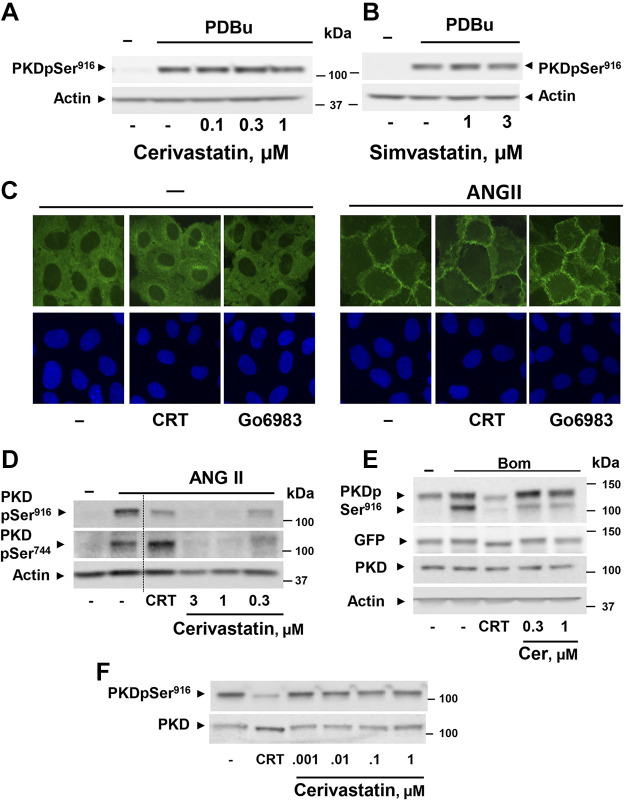
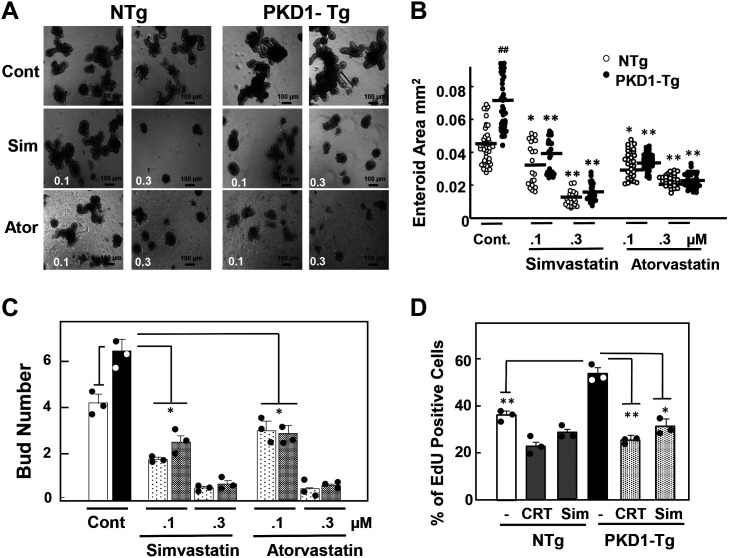
We examined the impact of statins on protein kinase D (PKD) activation by G protein-coupled receptor (GPCR) agonists. Treatment of intestinal IEC-18 cells with cerivastatin inhibited PKD autophosphorylation at Ser916 induced by angiotensin II (ANG II) or vasopressin in a dose-dependent manner with half-maximal inhibition at 0.2 µM. Cerivastatin treatment inhibited PKD activation stimulated by these agonists for different times (5-60 min) and blunted HDAC5 phosphorylation, a substrate of PKD. Other lipophilic statins, including simvastatin, atorvastatin, and fluvastatin also prevented PKD activation in a dose-dependent manner. Using IEC-18 cell lines expressing PKD1 tagged with EGFP (enhanced green fluorescent protein), cerivastatin or simvastatin blocked GPCR-mediated PKD1-EGFP translocation to the plasma membrane and its subsequent nuclear accumulation. Similar results were obtained in IEC-18 cells expressing PKD3-EGFP. Mechanistically, statins inhibited agonist-dependent PKD activation rather than acting directly on PKD catalytic activity since exposure to cerivastatin or simvastatin did not impair PKD autophosphorylation or PKD1-EGFP membrane translocation in response to phorbol dibutyrate, which bypasses GPCRs and directly stimulates PKC and PKD. Furthermore, cerivastatin did not inhibit recombinant PKD activity determined via an in vitro kinase assay. Using enteroids generated from intestinal crypt-derived epithelial cells from PKD1 transgenic mice as a model of intestinal regeneration, we show that statins oppose PKD1-mediated increase in enteroid area, complexity (number of crypt-like buds), and DNA synthesis. Our results revealed a previously unappreciated inhibitory effect of statins on receptor-mediated PKD activation and in opposing the growth-promoting effects of PKD1 on intestinal epithelial cells.
Keywords: CRT0066101; angiotensin II; atorvastatin; cerivastatin; intestinal IEC-18 cells.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
Similar articles
-
Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: role in mitogenic signaling.Am J Physiol Cell Physiol. 2014 May 15;306(10):C961-71. doi: 10.1152/ajpcell.00048.2014. Epub 2014 Mar 19. Am J Physiol Cell Physiol. 2014. PMID: 24647541 Free PMC article.
-
PKD1 mediates negative feedback of PI3K/Akt activation in response to G protein-coupled receptors.PLoS One. 2013 Sep 9;8(9):e73149. doi: 10.1371/journal.pone.0073149. eCollection 2013. PLoS One. 2013. PMID: 24039875 Free PMC article.
-
Biphasic Regulation of Yes-associated Protein (YAP) Cellular Localization, Phosphorylation, and Activity by G Protein-coupled Receptor Agonists in Intestinal Epithelial Cells: A NOVEL ROLE FOR PROTEIN KINASE D (PKD).J Biol Chem. 2016 Aug 19;291(34):17988-8005. doi: 10.1074/jbc.M115.711275. Epub 2016 Jul 1. J Biol Chem. 2016. PMID: 27369082 Free PMC article.
-
New insights into the pharmacodynamic and pharmacokinetic properties of statins.Pharmacol Ther. 1999 Dec;84(3):413-28. doi: 10.1016/s0163-7258(99)00045-5. Pharmacol Ther. 1999. PMID: 10665838 Review.
-
Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update.Fundam Clin Pharmacol. 2005 Feb;19(1):117-25. doi: 10.1111/j.1472-8206.2004.00299.x. Fundam Clin Pharmacol. 2005. PMID: 15660968 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous