

The Hypersaline Archaeal Histones HpyA and HstA Are DNA Binding Proteins That Defy Categorization According to Commonly Used Functional Criteria
- PMID: 36779711
- PMCID: PMC10128011
- DOI: 10.1128/mbio.03449-22
The Hypersaline Archaeal Histones HpyA and HstA Are DNA Binding Proteins That Defy Categorization According to Commonly Used Functional Criteria
Abstract
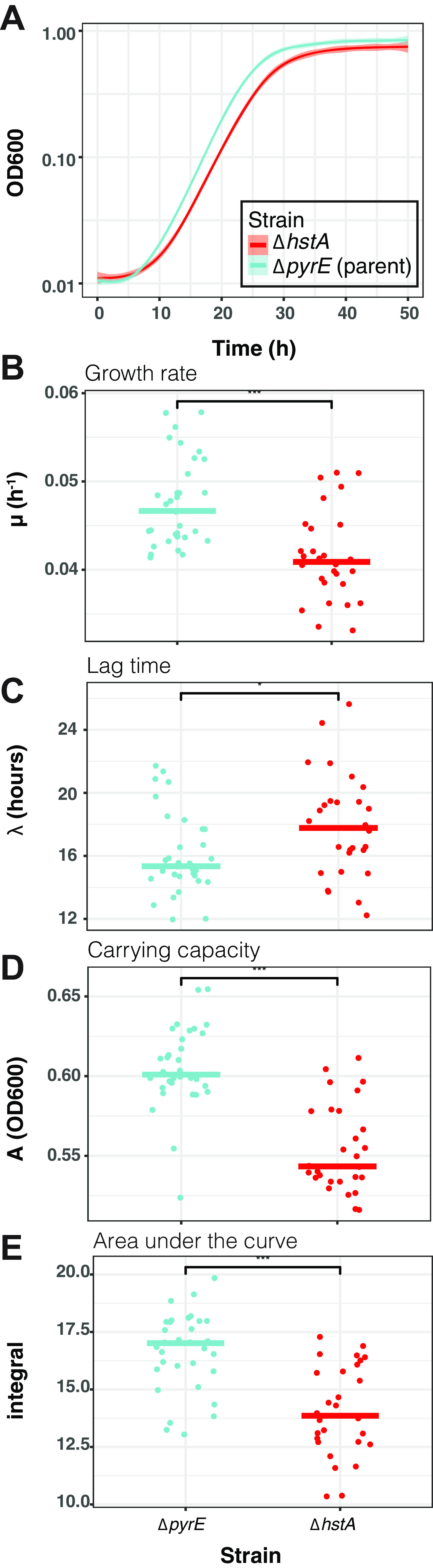
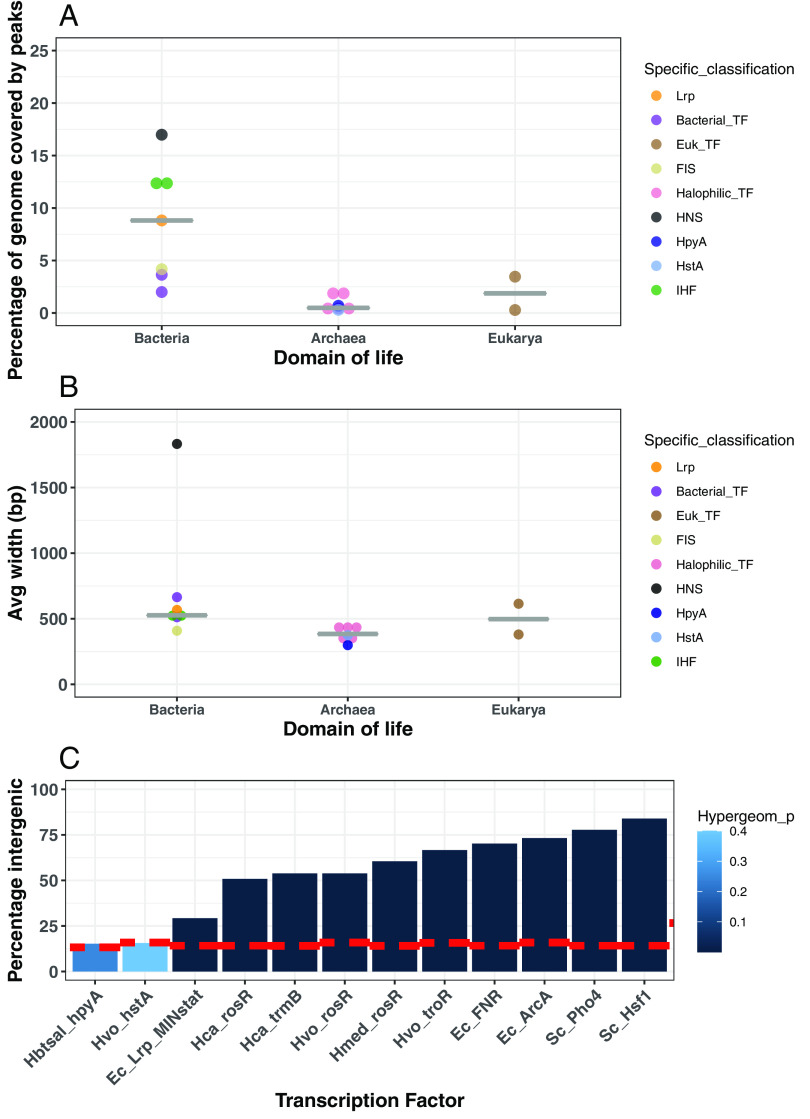
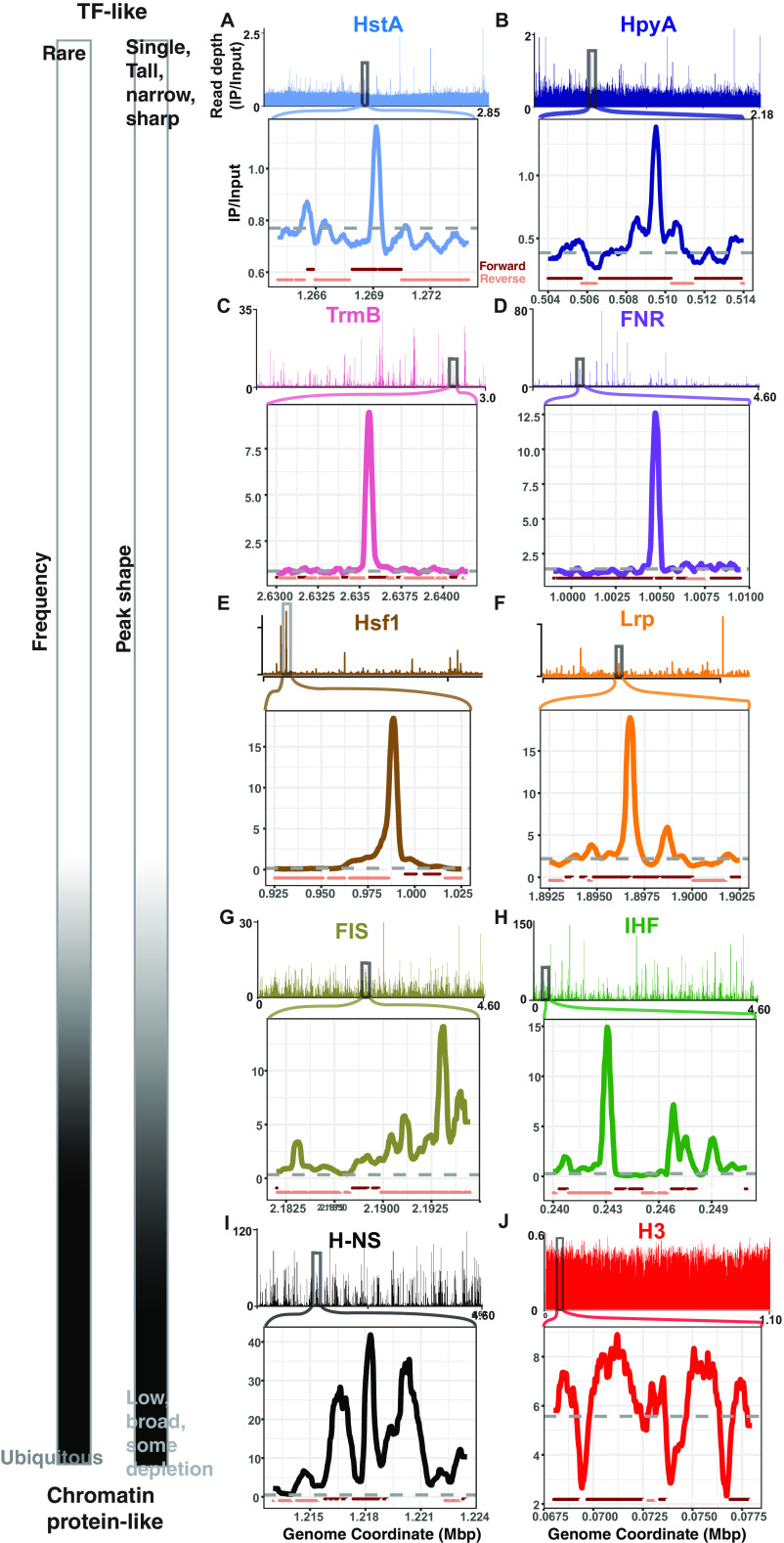
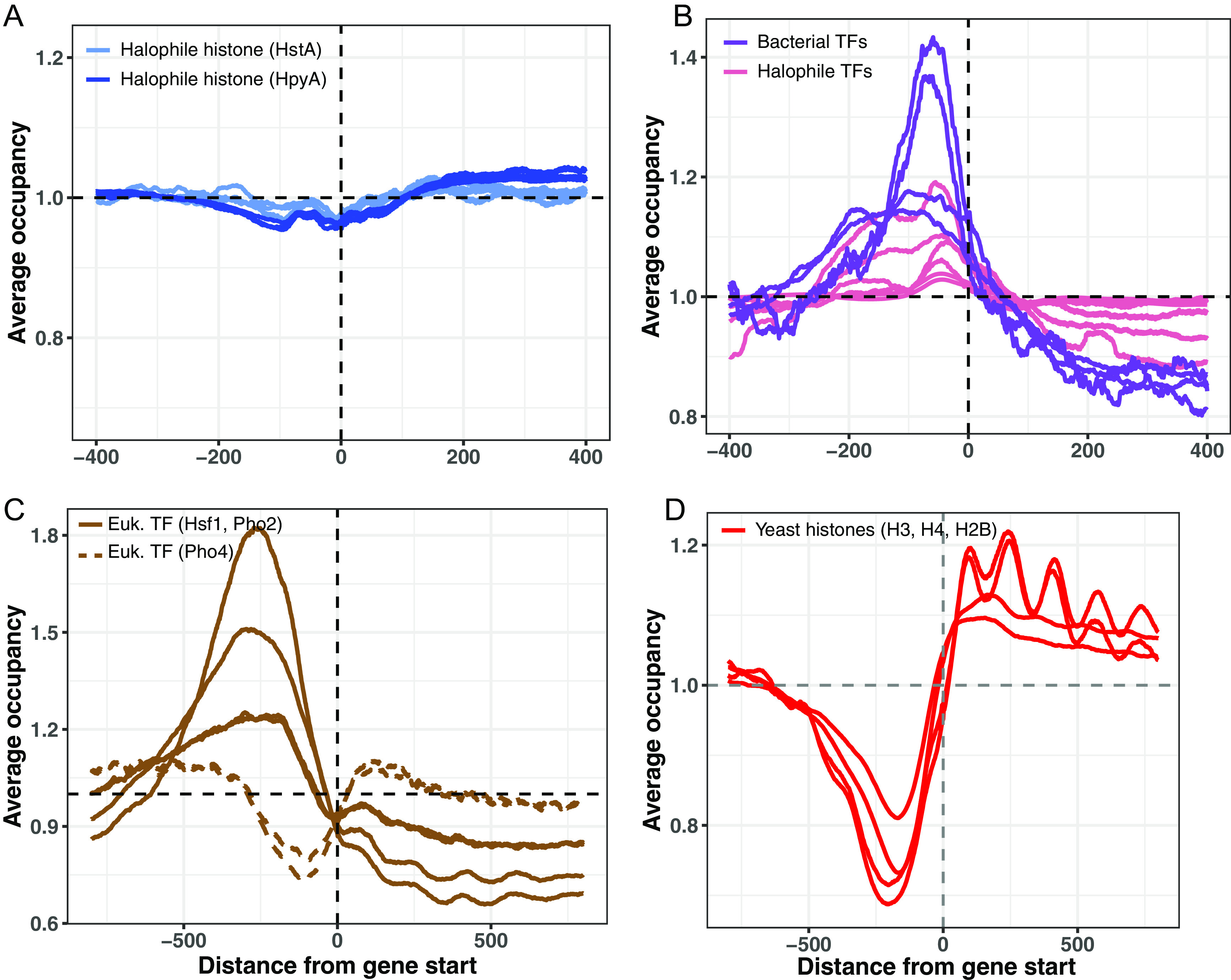
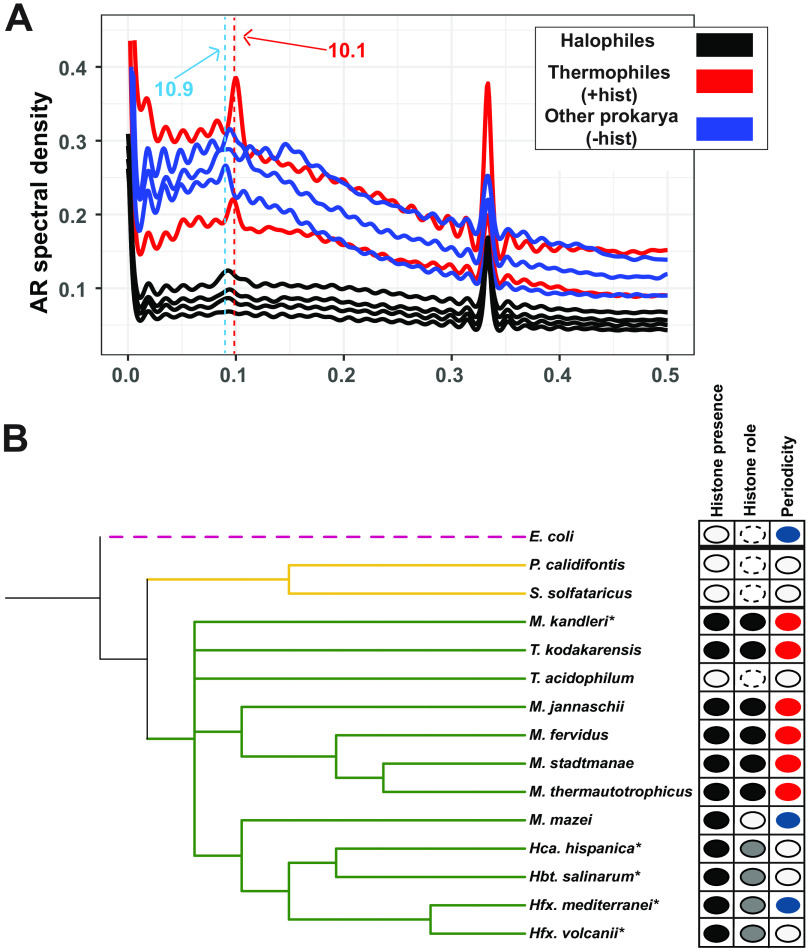
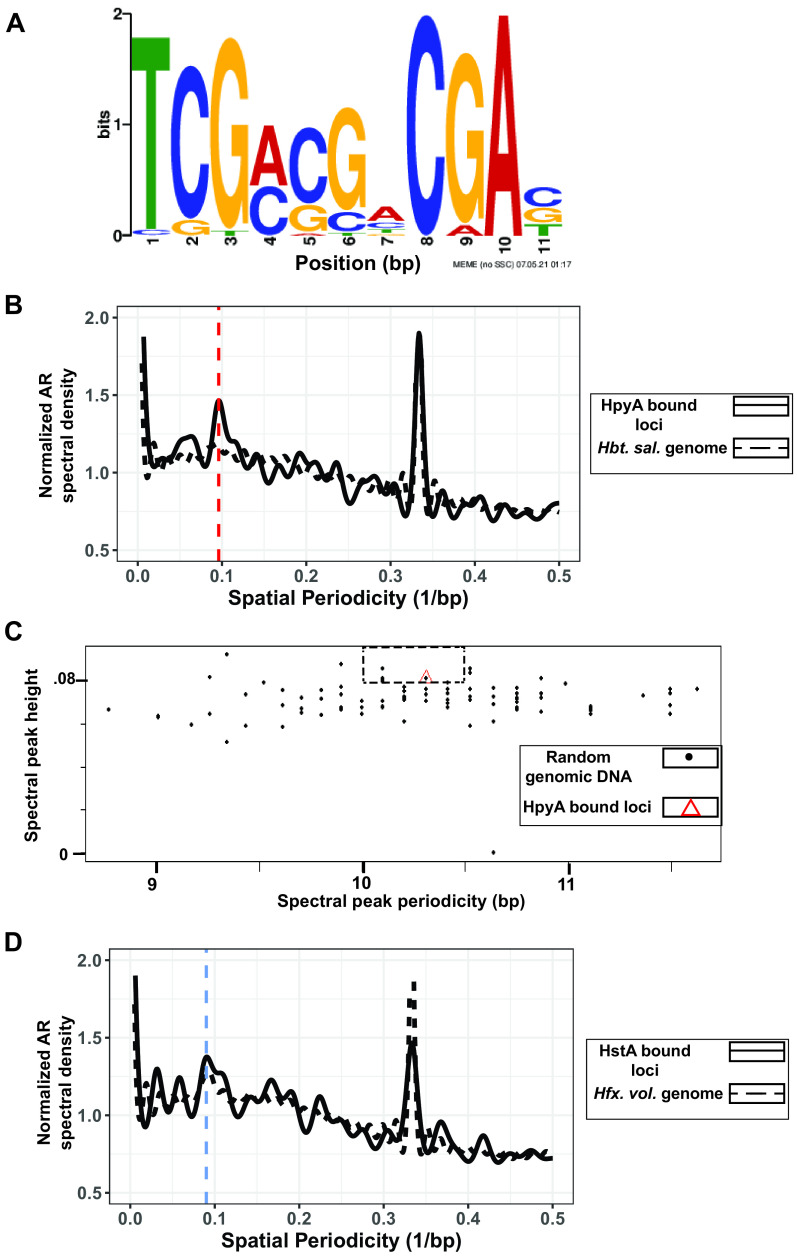
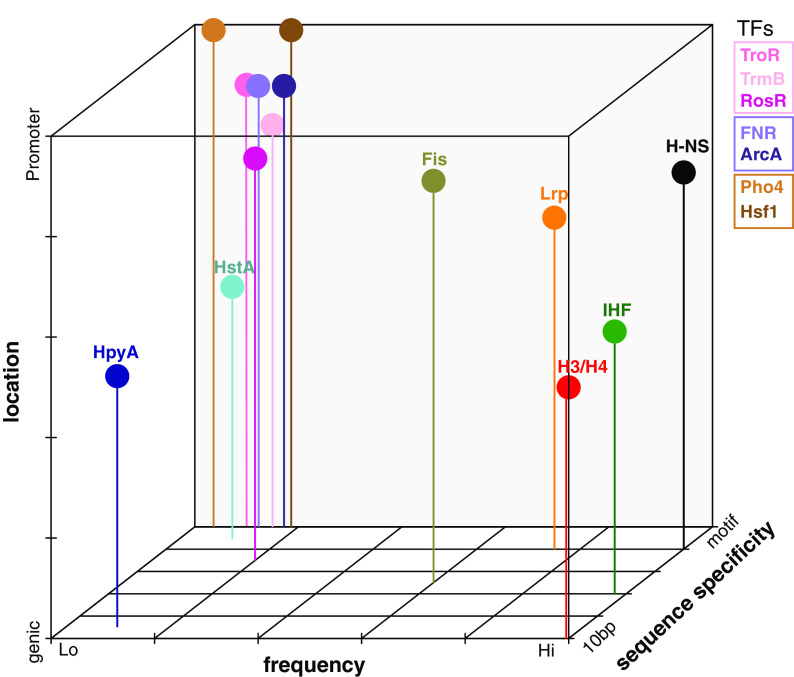
Histone proteins are found across diverse lineages of Archaea, many of which package DNA and form chromatin. However, previous research has led to the hypothesis that the histone-like proteins of high-salt-adapted archaea, or halophiles, function differently. The sole histone protein encoded by the model halophilic species Halobacterium salinarum, HpyA, is nonessential and expressed at levels too low to enable genome-wide DNA packaging. Instead, HpyA mediates the transcriptional response to salt stress. Here we compare the features of genome-wide binding of HpyA to those of HstA, the sole histone of another model halophile, Haloferax volcanii. hstA, like hpyA, is a nonessential gene. To better understand HpyA and HstA functions, protein-DNA binding data (chromatin immunoprecipitation sequencing [ChIP-seq]) of these halophilic histones are compared to publicly available ChIP-seq data from DNA binding proteins across all domains of life, including transcription factors (TFs), nucleoid-associated proteins (NAPs), and histones. These analyses demonstrate that HpyA and HstA bind the genome infrequently in discrete regions, which is similar to TFs but unlike NAPs, which bind a much larger genomic fraction. However, unlike TFs that typically bind in intergenic regions, HpyA and HstA binding sites are located in both coding and intergenic regions. The genome-wide dinucleotide periodicity known to facilitate histone binding was undetectable in the genomes of both species. Instead, TF-like and histone-like binding sequence preferences were detected for HstA and HpyA, respectively. Taken together, these data suggest that halophilic archaeal histones are unlikely to facilitate genome-wide chromatin formation and that their function defies categorization as a TF, NAP, or histone. IMPORTANCE Most cells in eukaryotic species-from yeast to humans-possess histone proteins that pack and unpack DNA in response to environmental cues. These essential proteins regulate genes necessary for important cellular processes, including development and stress protection. Although the histone fold domain originated in the domain of life Archaea, the function of archaeal histone-like proteins is not well understood relative to those of eukaryotes. We recently discovered that, unlike histones of eukaryotes, histones in hypersaline-adapted archaeal species do not package DNA and can act as transcription factors (TFs) to regulate stress response gene expression. However, the function of histones across species of hypersaline-adapted archaea still remains unclear. Here, we compare hypersaline histone function to a variety of DNA binding proteins across the tree of life, revealing histone-like behavior in some respects and specific transcriptional regulatory function in others.
Keywords: archaea; histones; regulation of gene expression.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
