

Recombineering: Genetic Engineering in Escherichia coli Using Homologous Recombination
- PMID: 36779782
- PMCID: PMC10037674
- DOI: 10.1002/cpz1.656
Recombineering: Genetic Engineering in Escherichia coli Using Homologous Recombination
Erratum in
-
Correction: Recombineering: Genetic Engineering in Escherichia coli Using Homologous Recombination.Curr Protoc. 2024 Nov;4(11):e70064. doi: 10.1002/cpz1.70064. Curr Protoc. 2024. PMID: 39540709 No abstract available.
Abstract
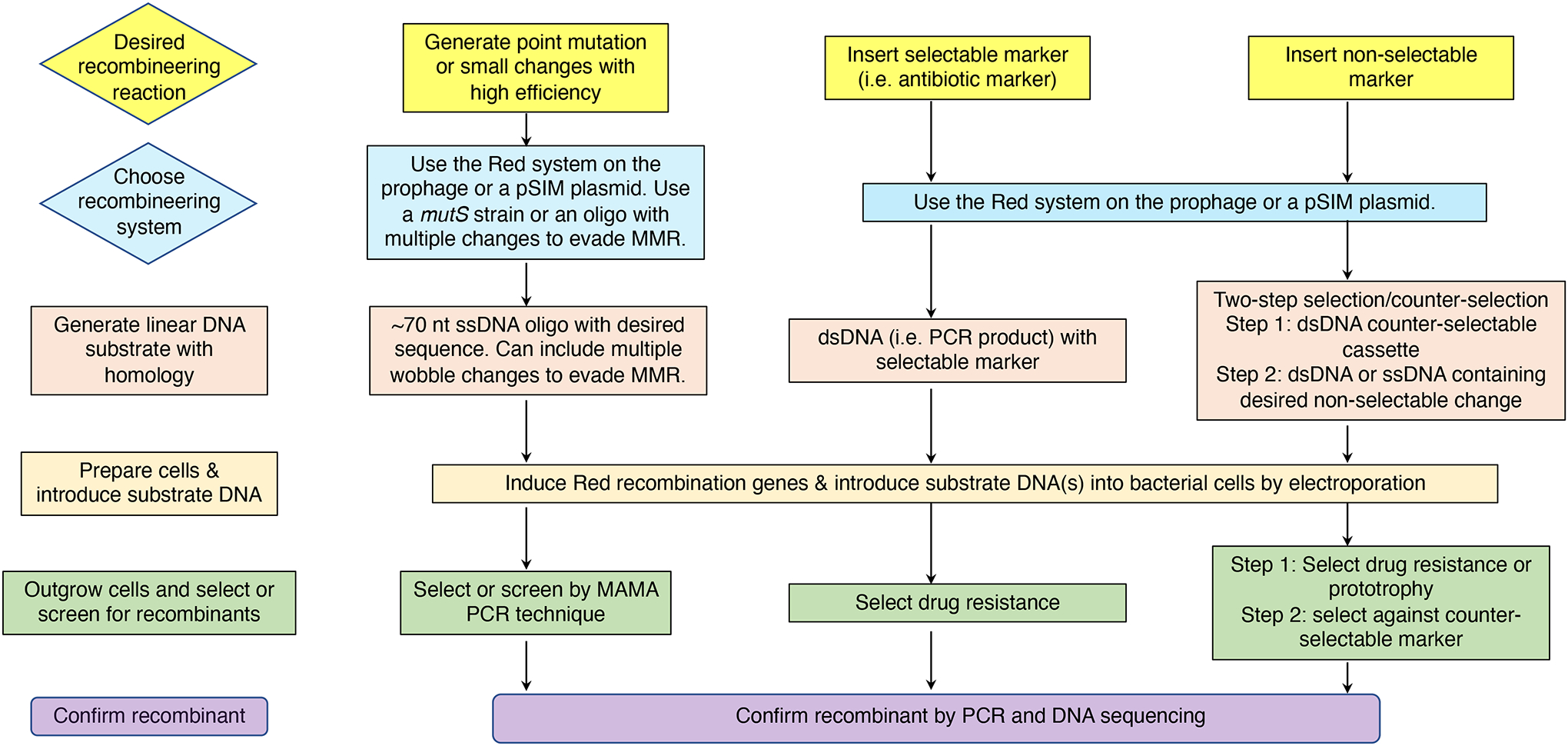
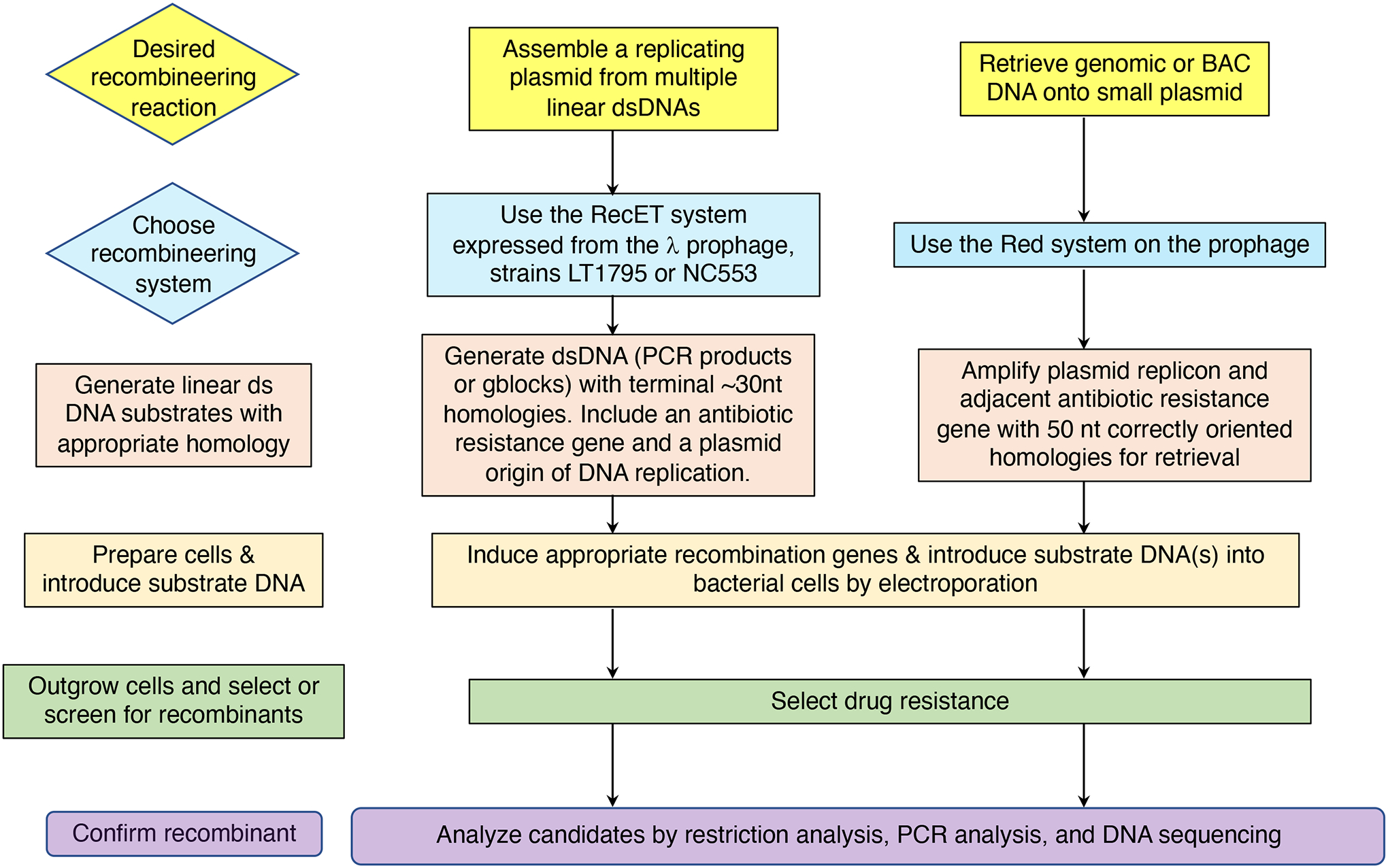
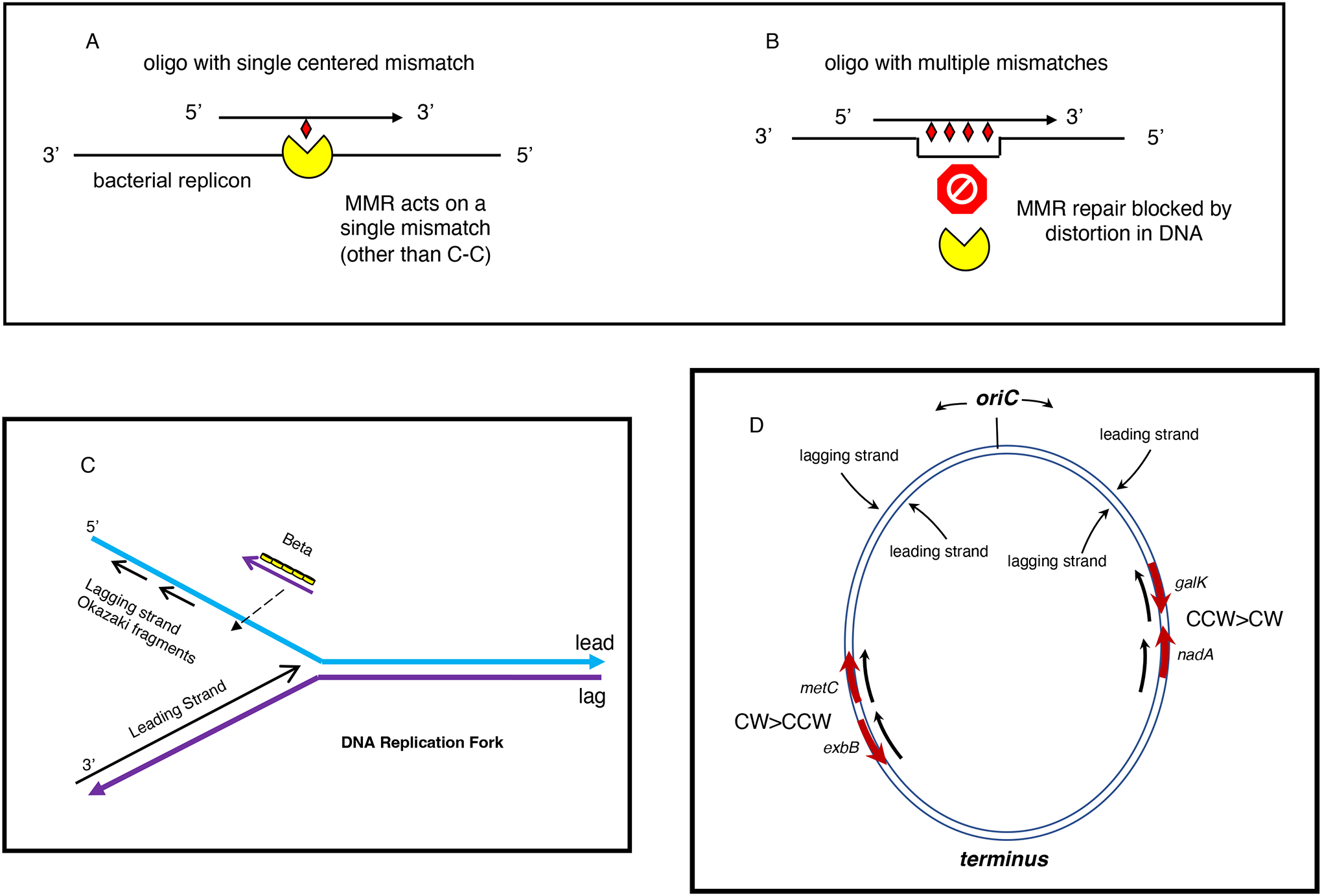
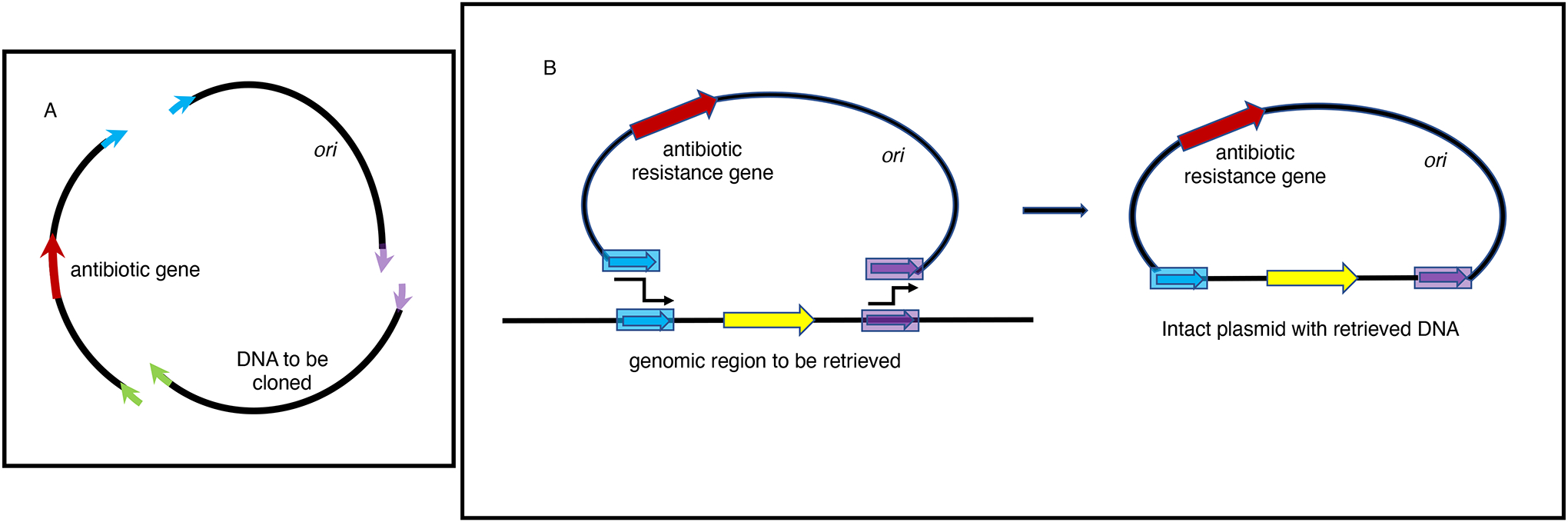
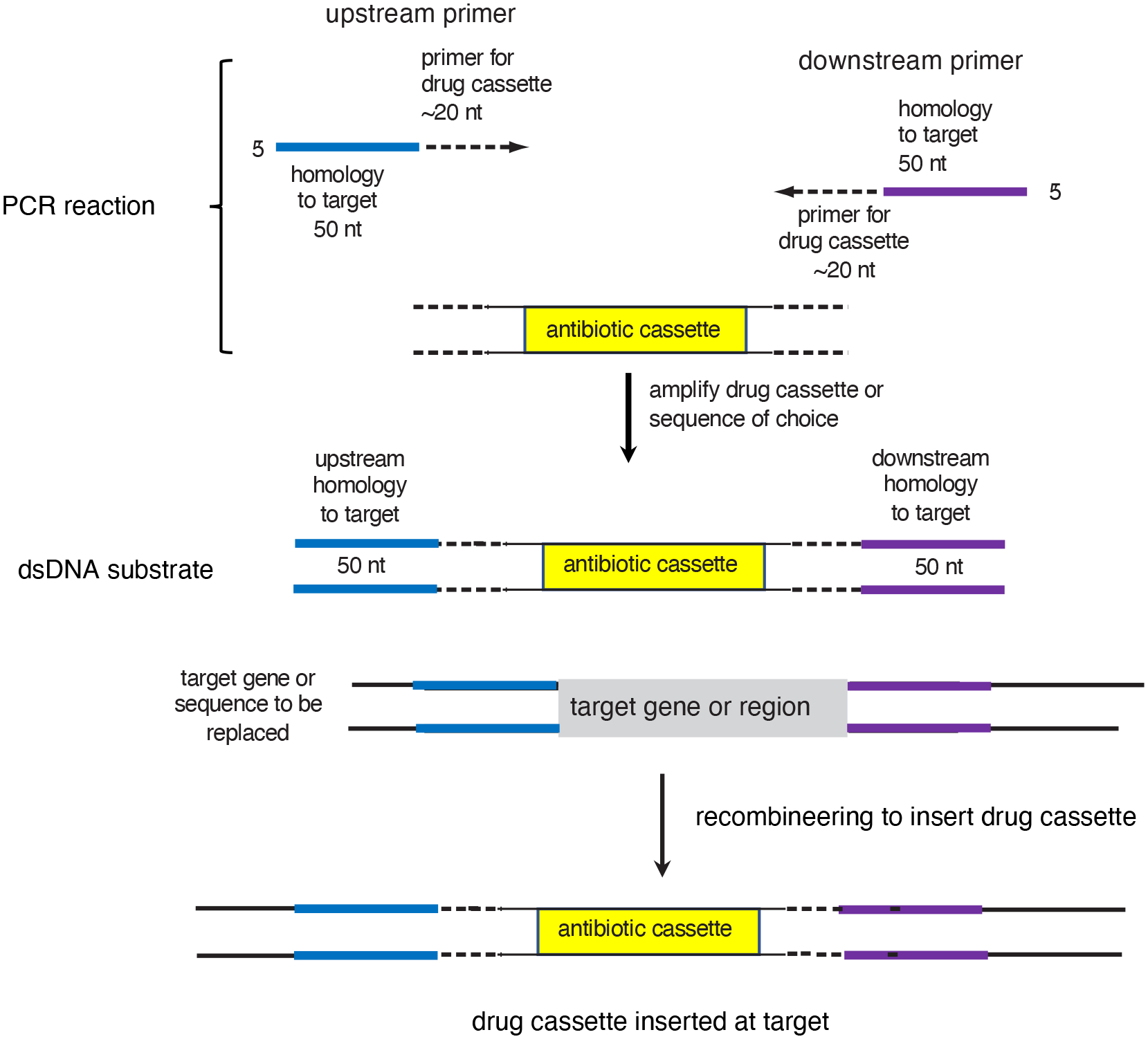
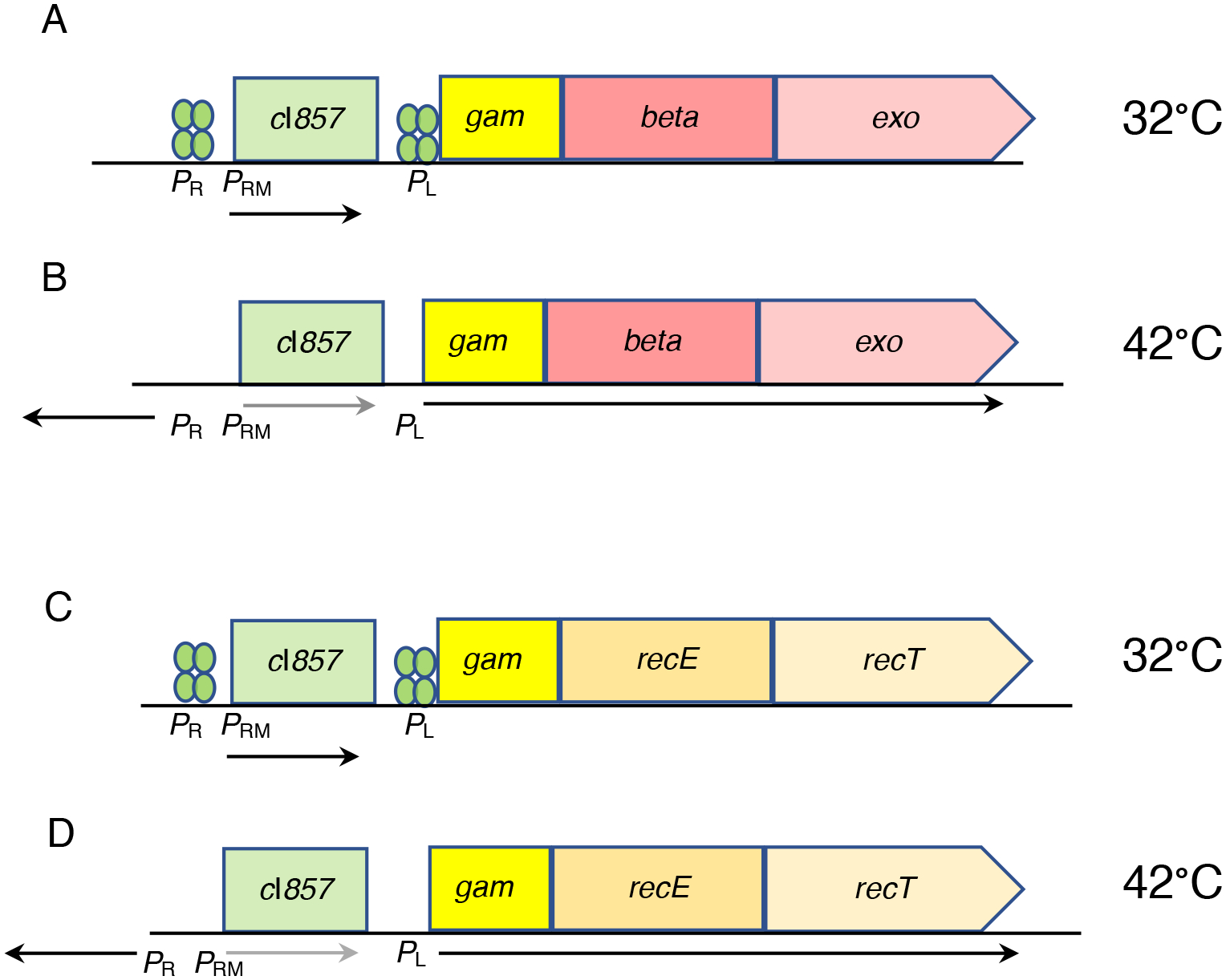
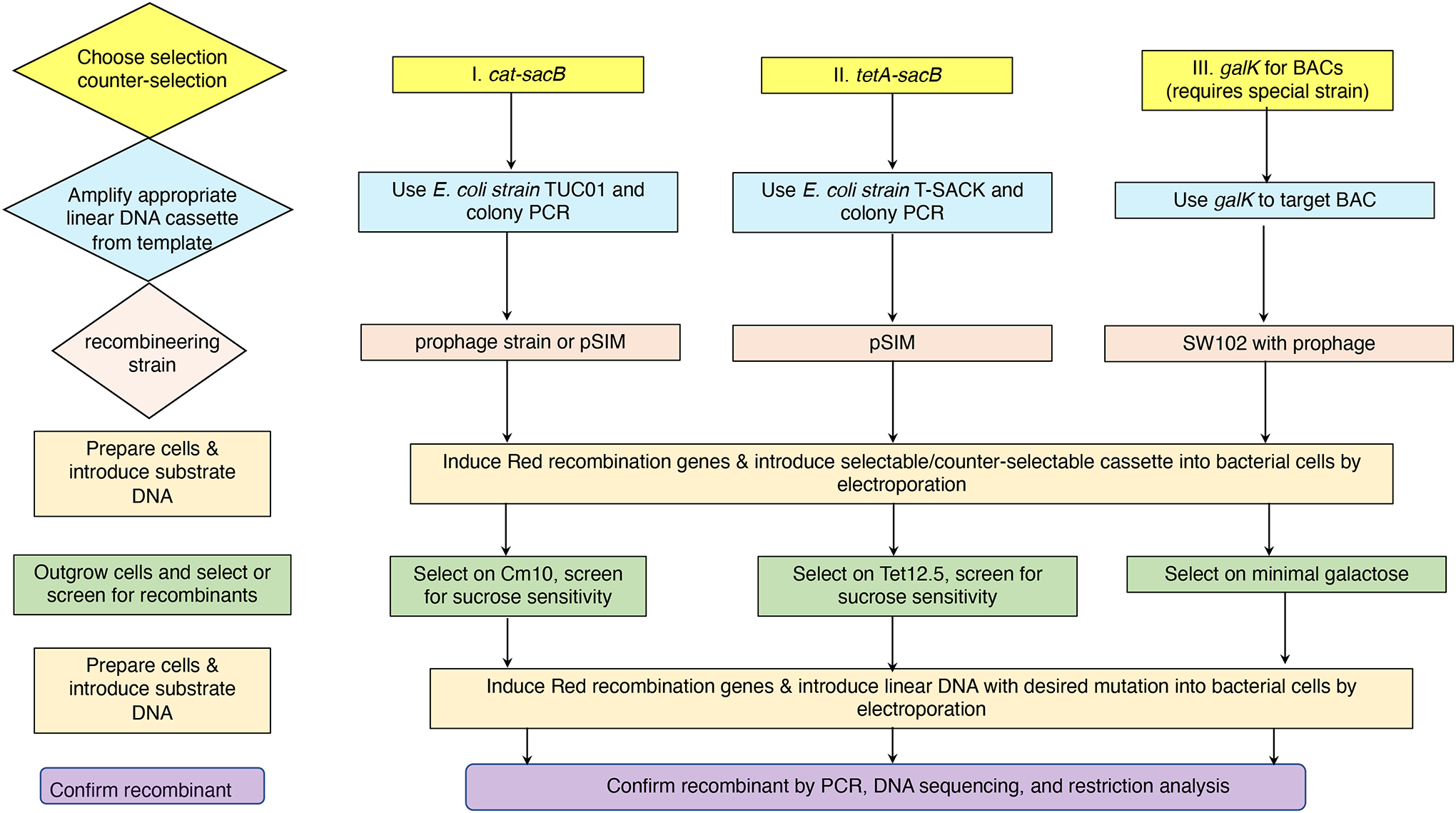
The bacterial chromosome and bacterial plasmids can be engineered in vivo by homologous recombination using either PCR products or synthetic double-stranded DNA (dsDNA) or single-stranded DNA as substrates. Multiple linear dsDNA molecules can be assembled into an intact plasmid. The technology of recombineering is possible because bacteriophage-encoded recombination proteins efficiently recombine sequences with homologies as short as 35 to 50 bases. Recombineering allows DNA sequences to be inserted or deleted without regard to the location of restriction sites and can also be used in combination with CRISPR/Cas targeting systems. © 2023 Wiley Periodicals LLC. This article has been contributed to by U.S. Government employees and their work is in the public domain in the USA. Basic Protocol: Making electrocompetent cells and transforming with linear DNA Support Protocol 1: Selection/counter-selections for genome engineering Support Protocol 2: Creating and screening for oligo recombinants by PCR Support Protocol 3: Other methods of screening for unselected recombinants Support Protocol 4: Curing recombineering plasmids containing a temperature-sensitive replication function Support Protocol 5: Removal of the prophage by recombineering Alternate Protocol 1: Using CRISPR/Cas9 as a counter-selection following recombineering Alternate Protocol 2: Assembly of linear dsDNA fragments into functional plasmids Alternate Protocol 3: Retrieval of alleles onto a plasmid by gap repair Alternate Protocol 4: Modifying multicopy plasmids with recombineering Support Protocol 6: Screening for unselected plasmid recombinants Alternate Protocol 5: Recombineering with an intact λ prophage Alternate Protocol 6: Targeting an infecting λ phage with the defective prophage strains.
Keywords: Escherichia coli; Rac prophage RecET; bacteriophage λ; homologous recombination; recombineering; λ Red system.
© 2023 Wiley Periodicals LLC. This article has been contributed to by U.S. Government employees and their work is in the public domain in the USA.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT:
The authors declare no conflict of interest.
Figures
Similar articles
-
Recombineering: genetic engineering in bacteria using homologous recombination.Curr Protoc Mol Biol. 2014 Apr 14;106:1.16.1-1.16.39. doi: 10.1002/0471142727.mb0116s106. Curr Protoc Mol Biol. 2014. PMID: 24733238 Review.
-
Recombineering in Non-Model Bacteria.Curr Protoc. 2022 Dec;2(12):e605. doi: 10.1002/cpz1.605. Curr Protoc. 2022. PMID: 36546891 Free PMC article.
-
Recombineering: genetic engineering in bacteria using homologous recombination.Curr Protoc Mol Biol. 2007 Apr;Chapter 1:Unit 1.16. doi: 10.1002/0471142727.mb0116s78. Curr Protoc Mol Biol. 2007. PMID: 18265390 Review.
-
Examining a DNA Replication Requirement for Bacteriophage λ Red- and Rac Prophage RecET-Promoted Recombination in Escherichia coli.mBio. 2016 Sep 13;7(5):e01443-16. doi: 10.1128/mBio.01443-16. mBio. 2016. PMID: 27624131 Free PMC article.
-
Modifying bacteriophage lambda with recombineering.Methods Mol Biol. 2009;501:239-51. doi: 10.1007/978-1-60327-164-6_21. Methods Mol Biol. 2009. PMID: 19066825
Cited by
-
Nuclease genes occupy boundaries of genetic exchange between bacteriophages.bioRxiv [Preprint]. 2023 Mar 23:2023.03.23.533998. doi: 10.1101/2023.03.23.533998. bioRxiv. 2023. Update in: NAR Genom Bioinform. 2023 Aug 24;5(3):lqad076. doi: 10.1093/nargab/lqad076. PMID: 36993569 Free PMC article. Updated. Preprint.
-
Coselection of BAC for Escherichia coli chromosomal DNA multiplex automated genome engineering.Biotechnol Lett. 2024 Dec 26;47(1):14. doi: 10.1007/s10529-024-03554-4. Biotechnol Lett. 2024. PMID: 39725731
-
Beyond a few bases: methods for large DNA insertion and gene targeting in plants.Plant J. 2025 Mar;121(6):e70099. doi: 10.1111/tpj.70099. Plant J. 2025. PMID: 40121601 Free PMC article. Review.
-
CnRed: Efficient, Marker-free Genome Engineering of Cupriavidus necator H16 by Adapted Lambda Red Recombineering.ACS Synth Biol. 2025 Mar 21;14(3):842-854. doi: 10.1021/acssynbio.4c00757. Epub 2025 Feb 24. ACS Synth Biol. 2025. PMID: 39989320 Free PMC article.
-
Role of the antiparallel double-stranded filament form of FtsA in activating the Escherichia coli divisome.mBio. 2024 Aug 14;15(8):e0168724. doi: 10.1128/mbio.01687-24. Epub 2024 Jul 23. mBio. 2024. PMID: 39041810 Free PMC article.
References
INTERNET RESOURCES:
-
- https://redrecombineering.ncifcrf.gov/
-
This is the original Court laboratory recombineering website. Although no longer regularly updated, it contains valuable information about recombineering in E. coli.
-
- https://frederick.cancer.gov/resources/repositories/Brb/#/recombineering... .
-
This link to the Biological Resources Branch at NCI-Frederick is used for strain requests, and also contains recombineering information.
-
- https://ecocyc.org .
-
As stated on the EcoCyc website, “EcoCyc is a scientific database for the bacterium Escherichia coli K-12 MG1655. The EcoCyc project performs literature-based curation of its genome, and of transcriptional regulation, transporters, and metabolic pathways. EcoCyc is part of the larger
BioCyc collection of thousands of Pathway/Genome Databases for sequenced genomes.”
Literature Cited
-
- Arber W, Enquist L, Hohn B, Murray N, & Murray K (1983). Experimental methods for use with lambda. In Hendrix RW, Roberts JW, Stahl FW, & Weisberg RA (Eds.), Lambda II (pp. 433–466). Cold Spring Harbor, New York: Cold Spring Harbor Laboratory.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
