

Single-Cell Discovery and Multiomic Characterization of Therapeutic Targets in Multiple Myeloma
- PMID: 36779841
- PMCID: PMC10102848
- DOI: 10.1158/0008-5472.CAN-22-1769
Single-Cell Discovery and Multiomic Characterization of Therapeutic Targets in Multiple Myeloma
Abstract
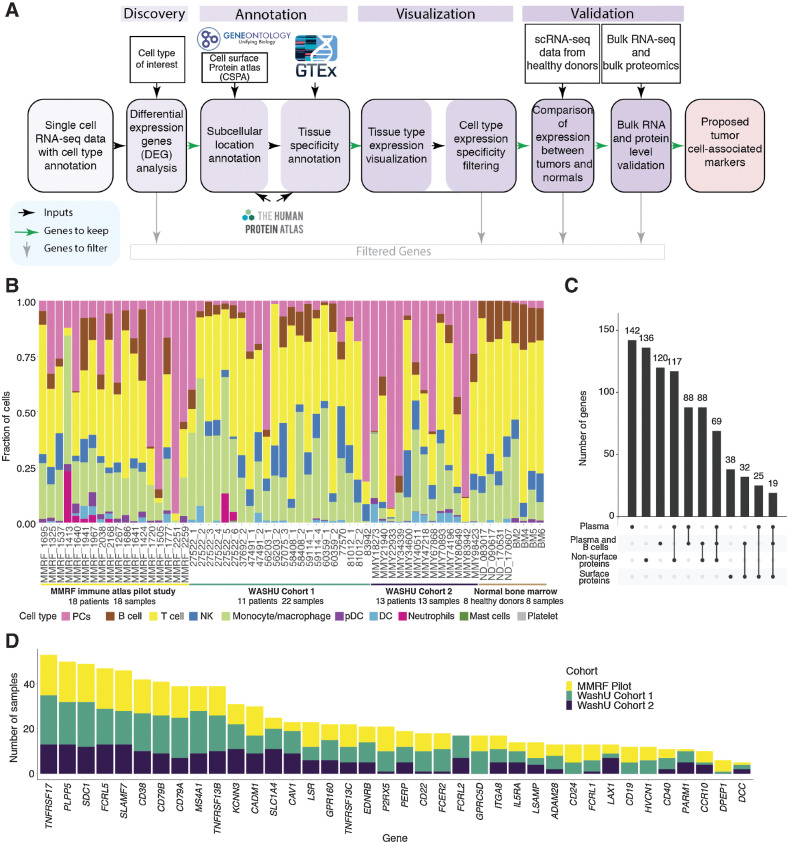
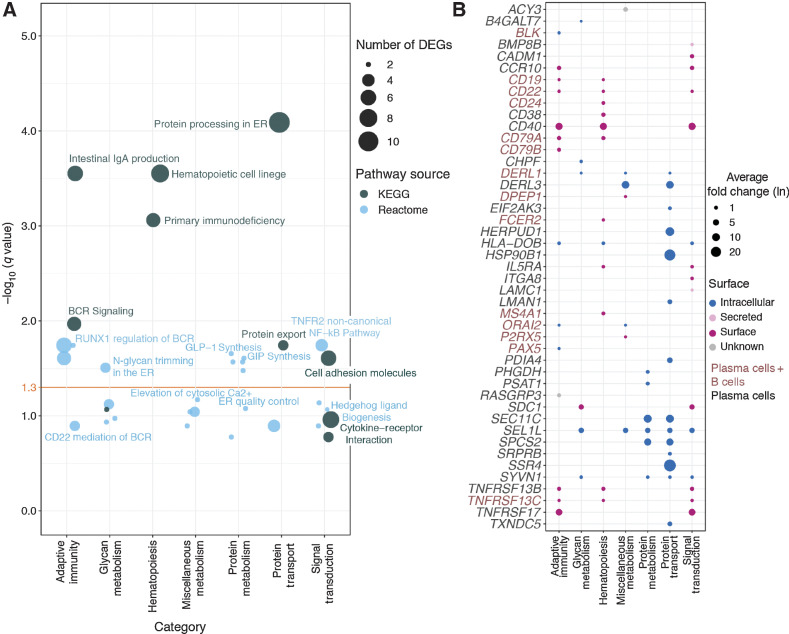
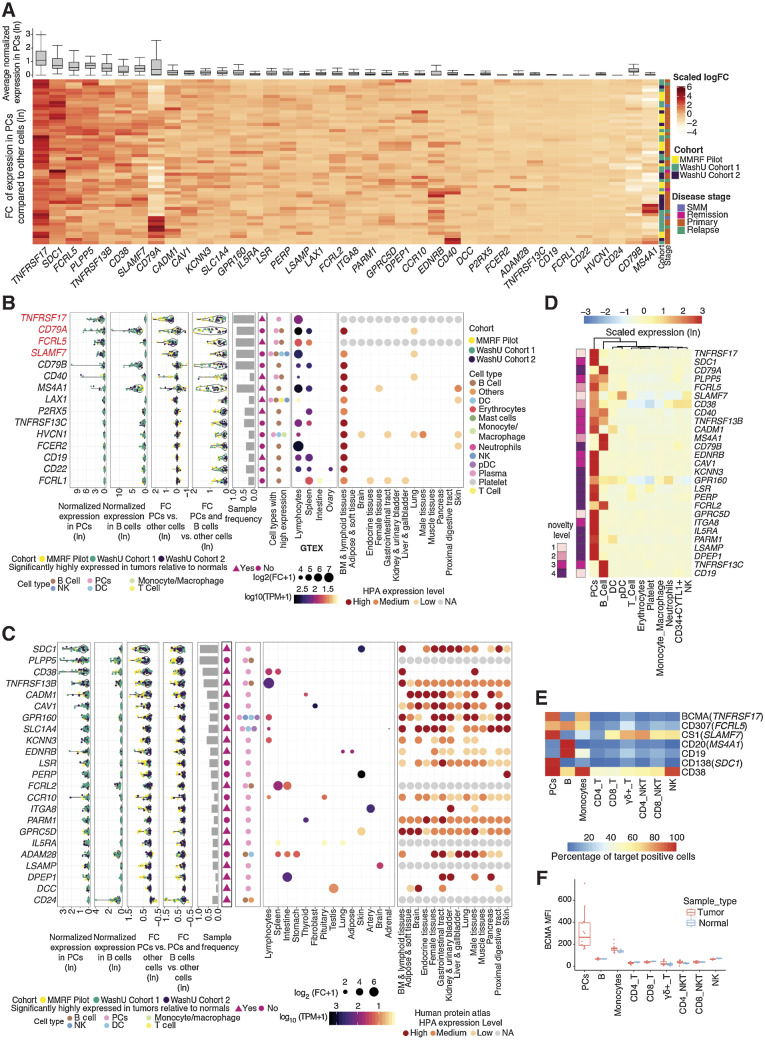
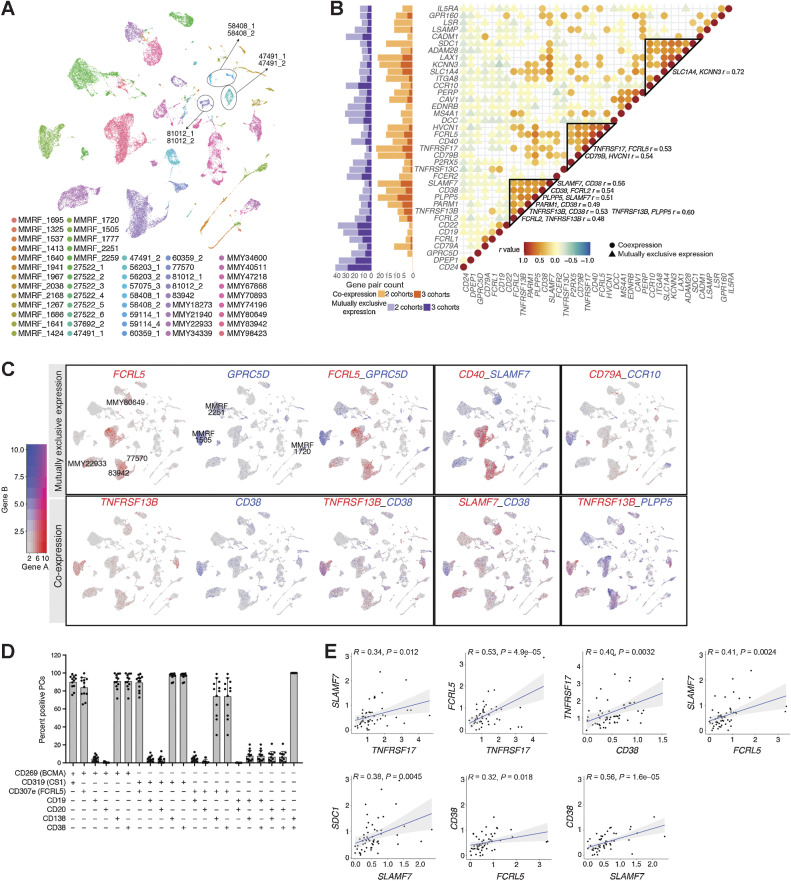
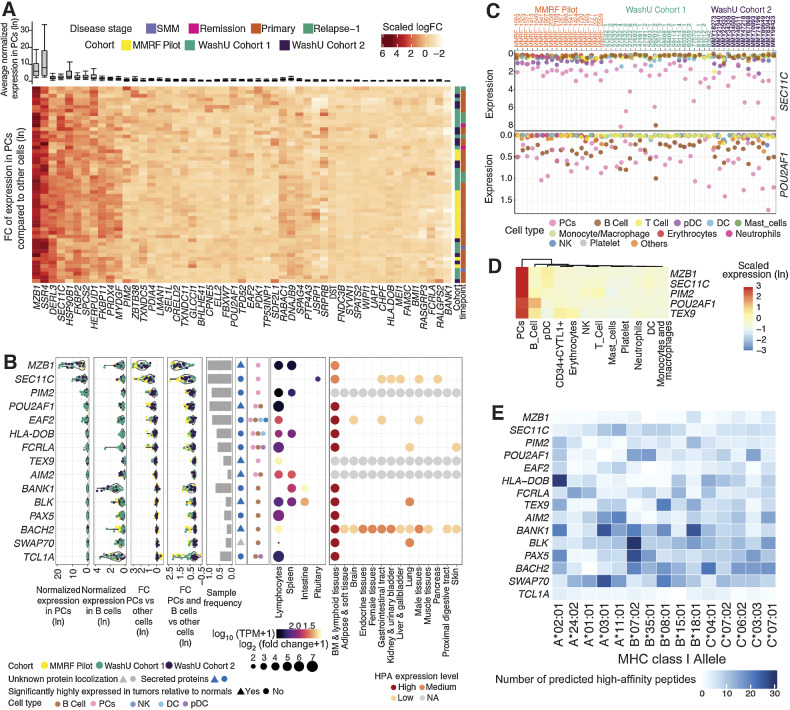
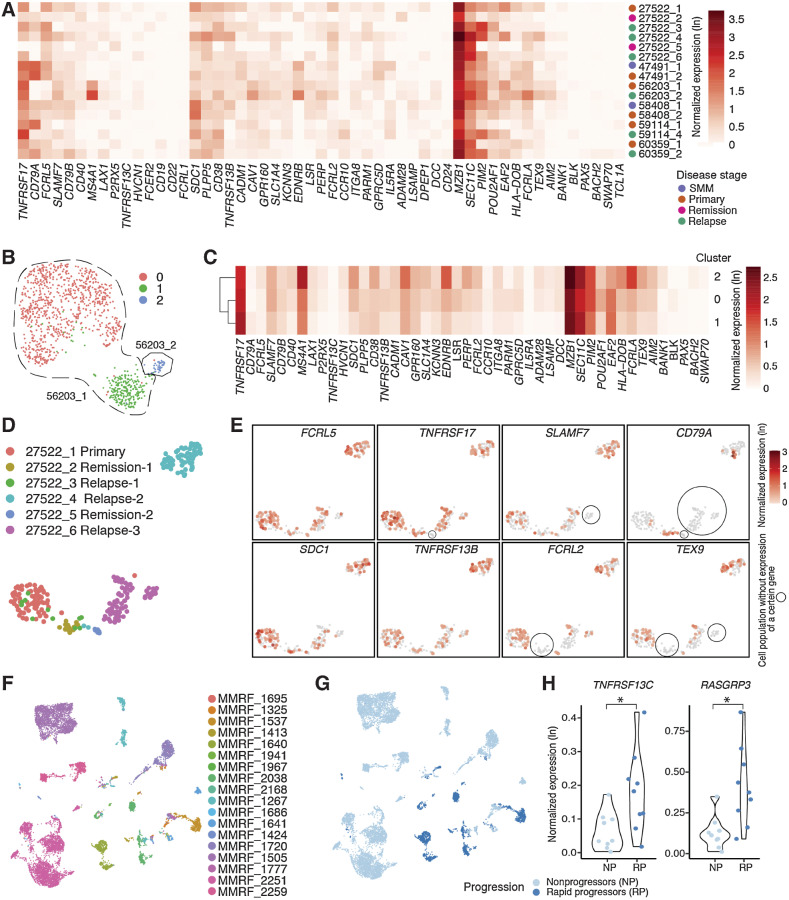
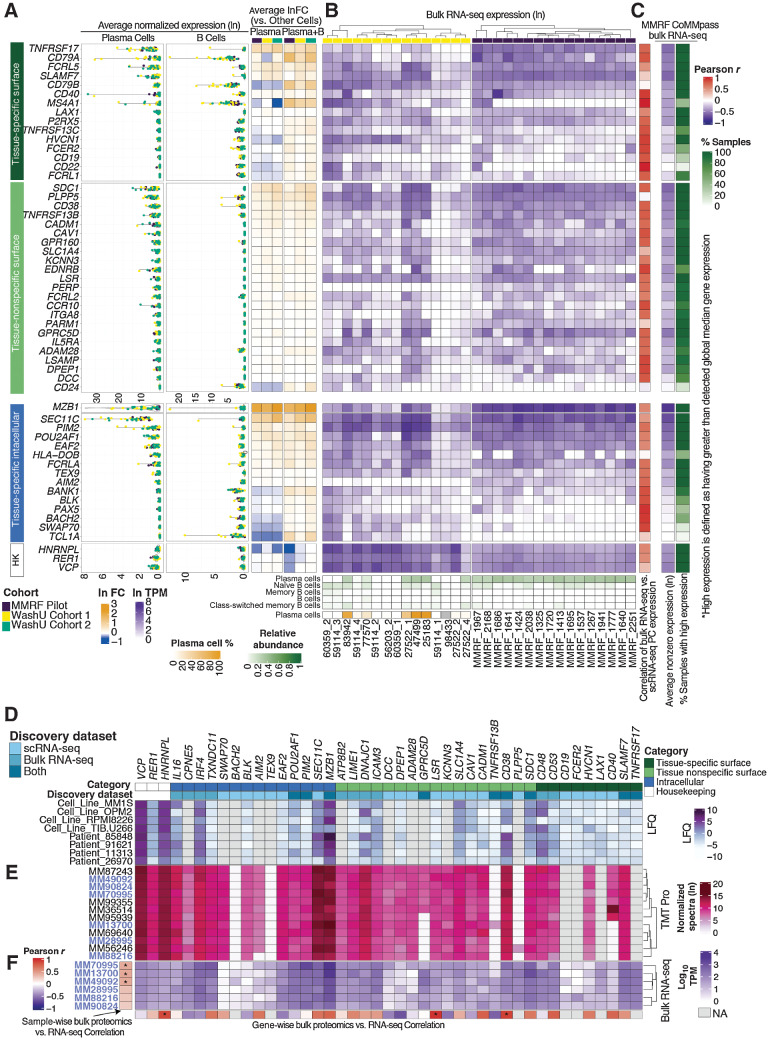
Multiple myeloma (MM) is a highly refractory hematologic cancer. Targeted immunotherapy has shown promise in MM but remains hindered by the challenge of identifying specific yet broadly representative tumor markers. We analyzed 53 bone marrow (BM) aspirates from 41 MM patients using an unbiased, high-throughput pipeline for therapeutic target discovery via single-cell transcriptomic profiling, yielding 38 MM marker genes encoding cell-surface proteins and 15 encoding intracellular proteins. Of these, 20 candidate genes were highlighted that are not yet under clinical study, 11 of which were previously uncharacterized as therapeutic targets. The findings were cross-validated using bulk RNA sequencing, flow cytometry, and proteomic mass spectrometry of MM cell lines and patient BM, demonstrating high overall concordance across data types. Independent discovery using bulk RNA sequencing reiterated top candidates, further affirming the ability of single-cell transcriptomics to accurately capture marker expression despite limitations in sample size or sequencing depth. Target dynamics and heterogeneity were further examined using both transcriptomic and immuno-imaging methods. In summary, this study presents a robust and broadly applicable strategy for identifying tumor markers to better inform the development of targeted cancer therapy.
Significance: Single-cell transcriptomic profiling and multiomic cross-validation to uncover therapeutic targets identifies 38 myeloma marker genes, including 11 transcribing surface proteins with previously uncharacterized potential for targeted antitumor therapy.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Gogishvili T, Danhof S, Prommersberger S, Rydzek J, Schreder M, Brede C, et al. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood 2017;130:2838–47. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
